アドセトリス点滴静注用50mg 発売中
進⾏期ホジキンリンパ腫


臨床成績
「警告・禁忌を含む使⽤上の注意」につきましては、「電子添文」をご参照ください。
⽇本が参加した国際共同第Ⅲ相⾮盲検2群⽐較試験(検証試験)
ブレンツキシマブ ベドチンの国際共同第Ⅲ相試験成績①(2018年9⽉21⽇承認、CTD 2.7.6.1、2.7.3.3、2.7.4.2)(承認審査時評価資料)
Connors JM, et al. : N Engl J Med. 2018; 378(4) : 331-344.
本試験は、Seattle Genetics 社(現・Seagen 社)とMillennium Pharmaceuticals 社(現・武⽥薬品⼯業株式会社)の⽀援により実施された。
本論⽂の著者のうちそれぞれ2名、4名は同社の社員で、試験計画、解析、執筆等の⽀援を受けている。
著者に同社より研究⽀援、謝礼⾦等を受領している者が含まれる。
Connors JM, et al. : N Engl J Med. 2018; 378(4) : 331-344. Supplementary appendix.
試験概要を確認する
試験概要
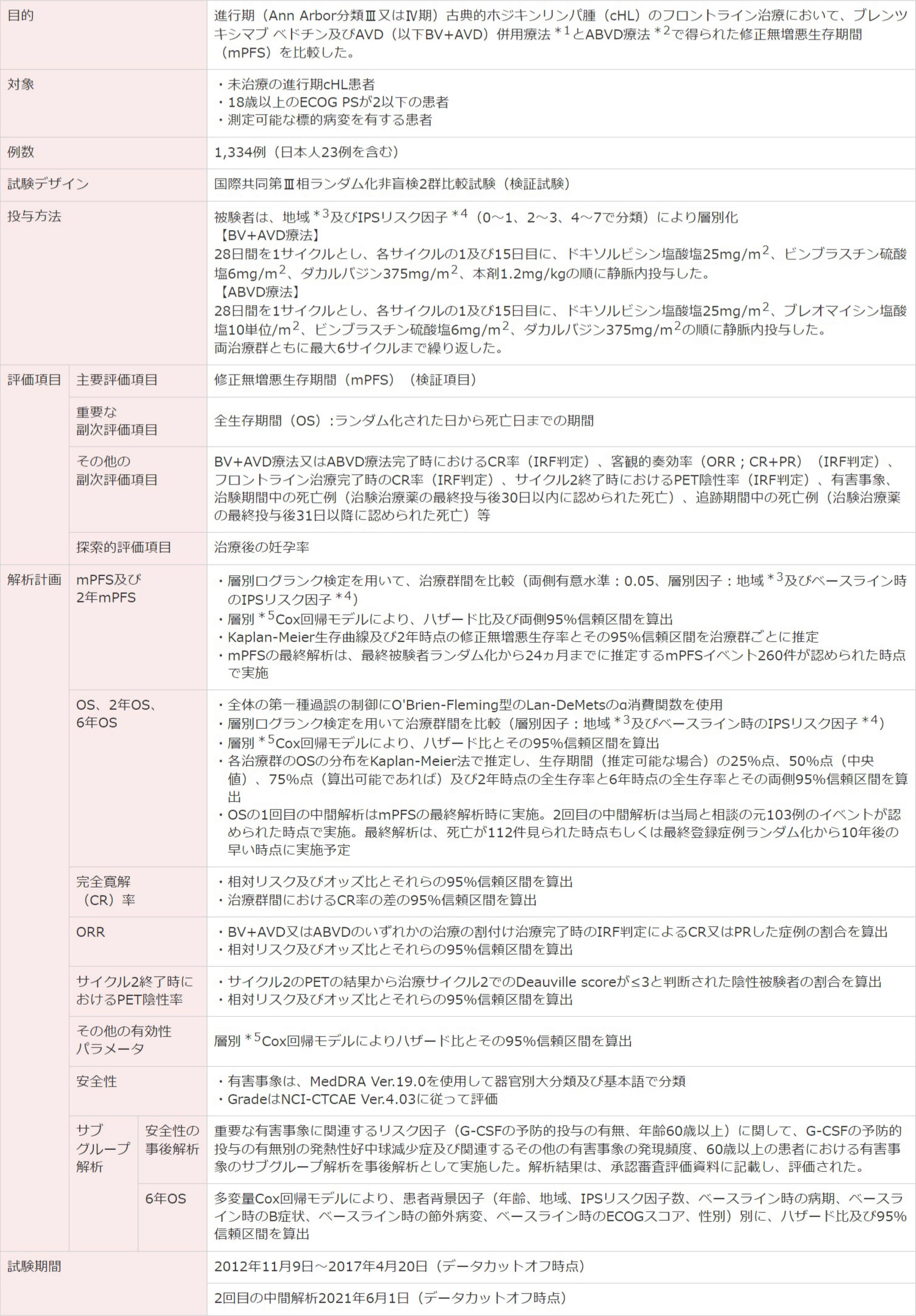
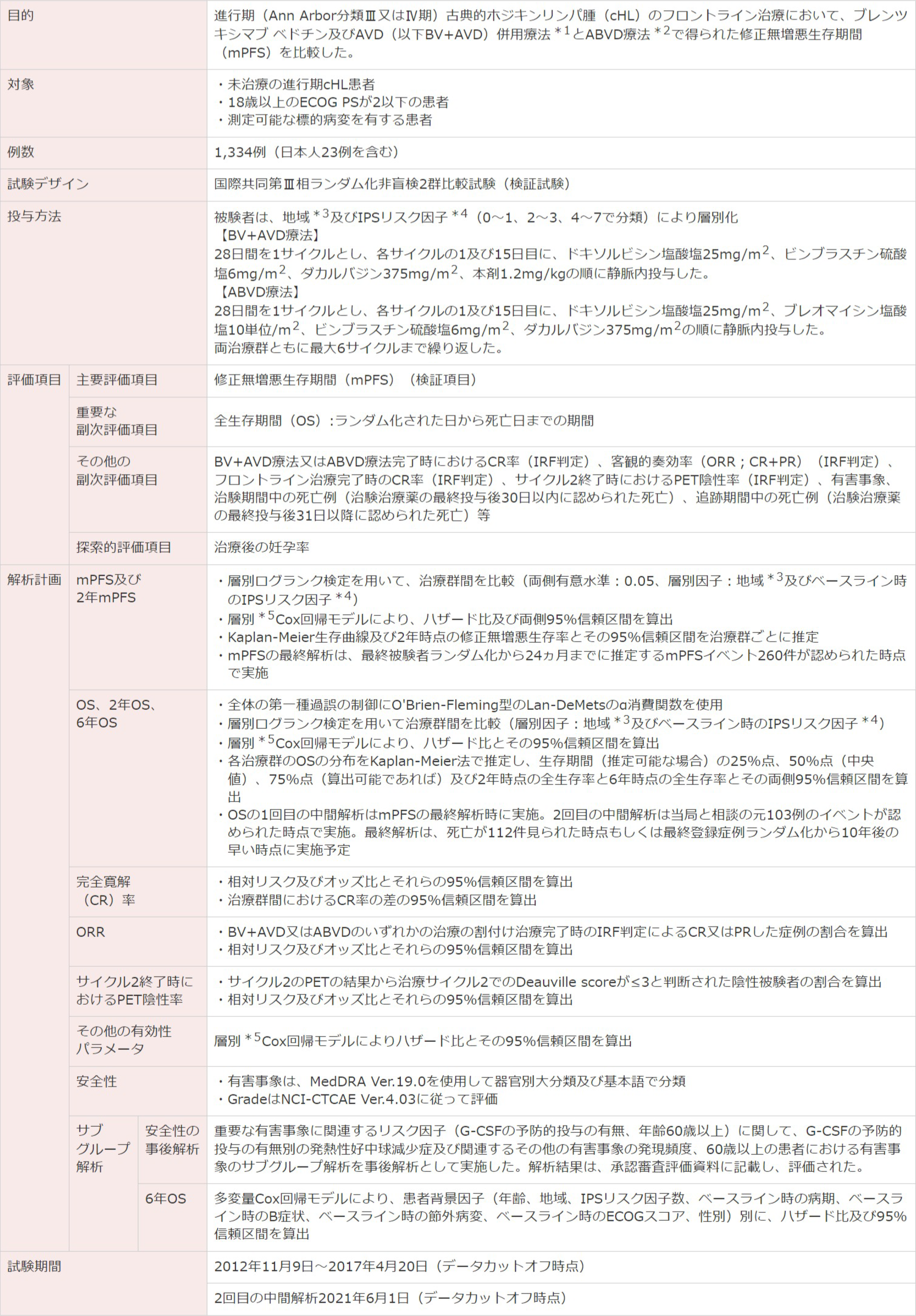
試験デザイン
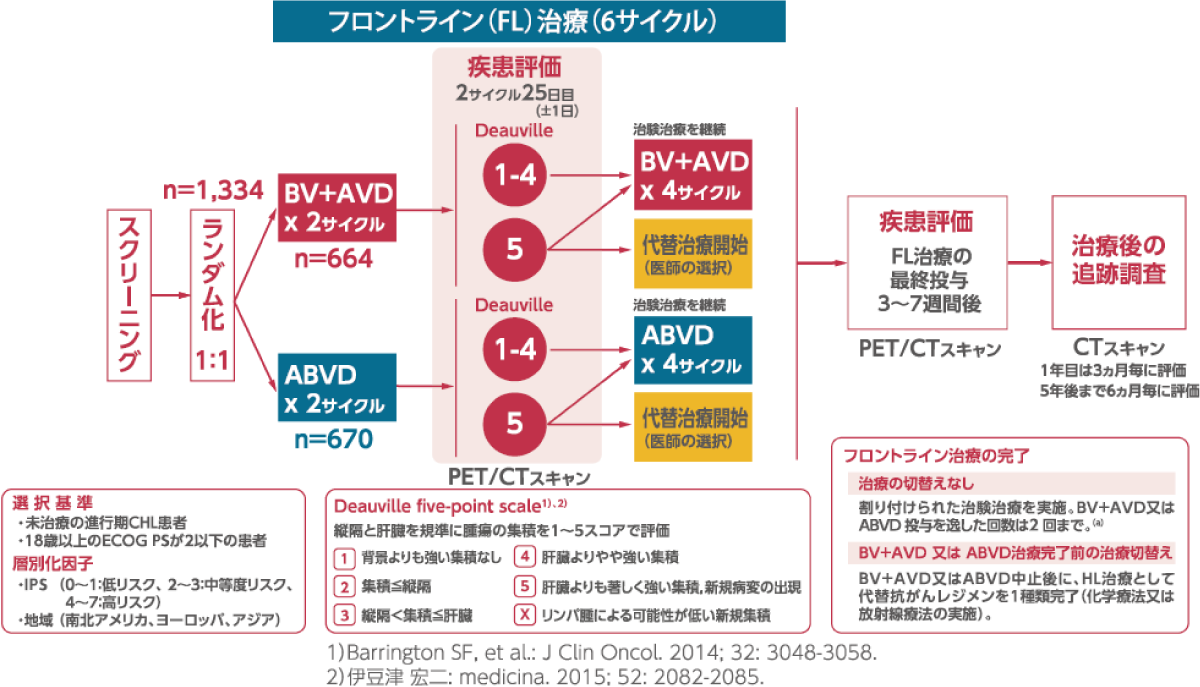
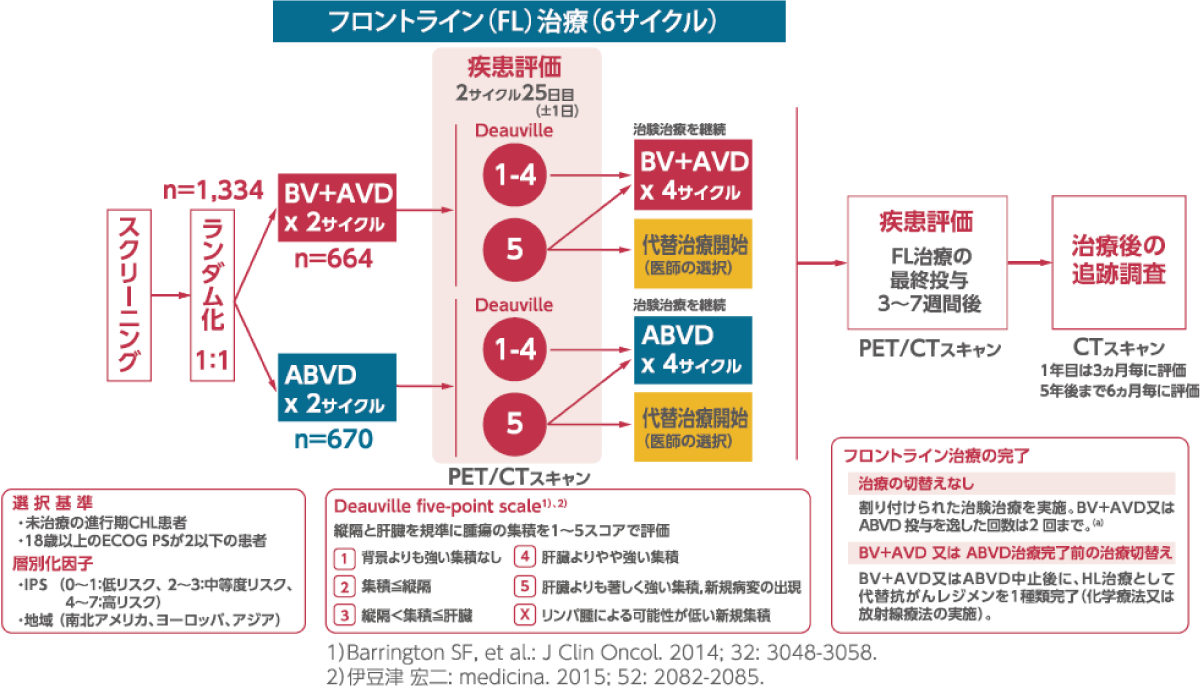
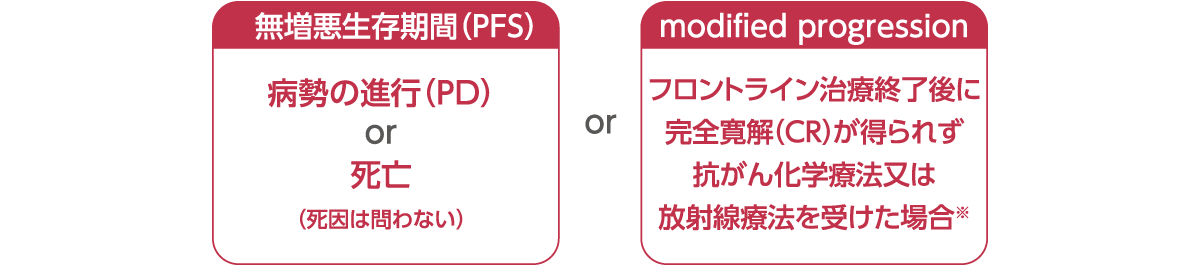
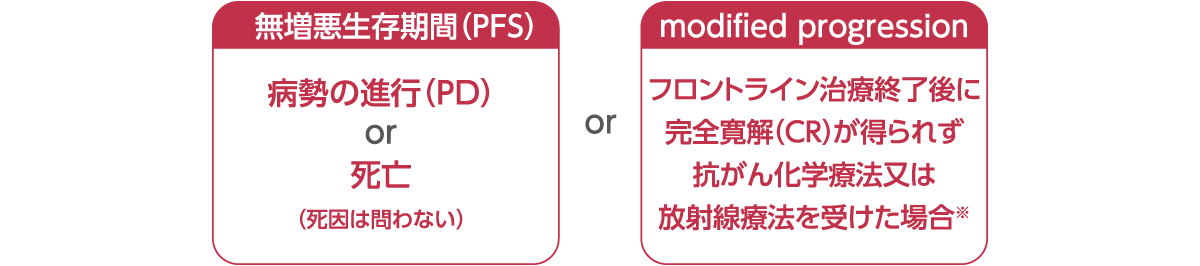
[主要評価項目]修正無増悪生存期間(mPFS)
ECOG PS:米国東海岸がん臨床試験グループのパフォーマンスステータス。IRF:中央判定委員会。PR : 部分寛解。IPS:国際予後スコア。
*1 本剤、ドキソルビシン塩酸塩、ビンブラスチン硫酸塩、ダカルバジン併用群。
*2 ドキソルビシン塩酸塩、ブレオマイシン塩酸塩、ビンブラスチン硫酸塩、ダカルバジン併用群。
*3 地域:南北アメリカ、ヨーロッパ、アジア。
*4 IPSリスク因子: 1)男性、2)45歳以上、3)Ann Arbor 分類Ⅳ期、4)ヘモグロビン値<10.5g/dL、5)白血球数≧15,000/mm3、6)リンパ球数<600/mm3又は白血球分画<8%、7)血清アルブミン値<4.0g/dLを予後不良因子として因子数により0-1、2-3、4-7に分類
(Hasenclever D, et al. : N Engl J Med. 1998; 339 : 1506-1514.)。
*5 層別因子:地域、ベースライン時のIPSリスク因子。
(a)「 投与を逸した」とはBV+AVD又はABVDレジメンの構成薬剤全てを投与しなかった場合を指す。BV+AVD又はABVDレジメンを構成する薬剤の一部を投与しなかった場合(肺毒性によりブレオマイシン塩酸塩の投与を中止するなど)は投与を逸したとはみなさない。
※なおイベント発生日は、フロントライン治療終了後の初回PETでCRが得られずDeauville score 3以上と判定された日とした。
ECOG PS:米国東海岸がん臨床試験グループのパフォーマンスステータス。IRF:中央判定委員会。PR : 部分寛解。IPS:国際予後スコア。
*1 本剤、ドキソルビシン塩酸塩、ビンブラスチン硫酸塩、ダカルバジン併用群。
*2 ドキソルビシン塩酸塩、ブレオマイシン塩酸塩、ビンブラスチン硫酸塩、ダカルバジン併用群。
*3 地域:南北アメリカ、ヨーロッパ、アジア。
*4 IPSリスク因子: 1)男性、2)45歳以上、3)Ann Arbor 分類Ⅳ期、4)ヘモグロビン値<10.5g/dL、5)白血球数≧15,000/mm3、6)リンパ球数<600/mm3又は白血球分画<8%、7)血清アルブミン値<4.0g/dLを予後不良因子として因子数により0-1、2-3、4-7に分類
(Hasenclever D, et al. : N Engl J Med. 1998; 339 : 1506-1514.)。
*5 層別因子:地域、ベースライン時のIPSリスク因子。
(a)「 投与を逸した」とはBV+AVD又はABVDレジメンの構成薬剤全てを投与しなかった場合を指す。BV+AVD又はABVDレジメンを構成する薬剤の一部を投与しなかった場合(肺毒性によりブレオマイシン塩酸塩の投与を中止するなど)は投与を逸したとはみなさない。
※なおイベント発生日は、フロントライン治療終了後の初回PETでCRが得られずDeauville score 3以上と判定された日とした。
試験結果を確認する
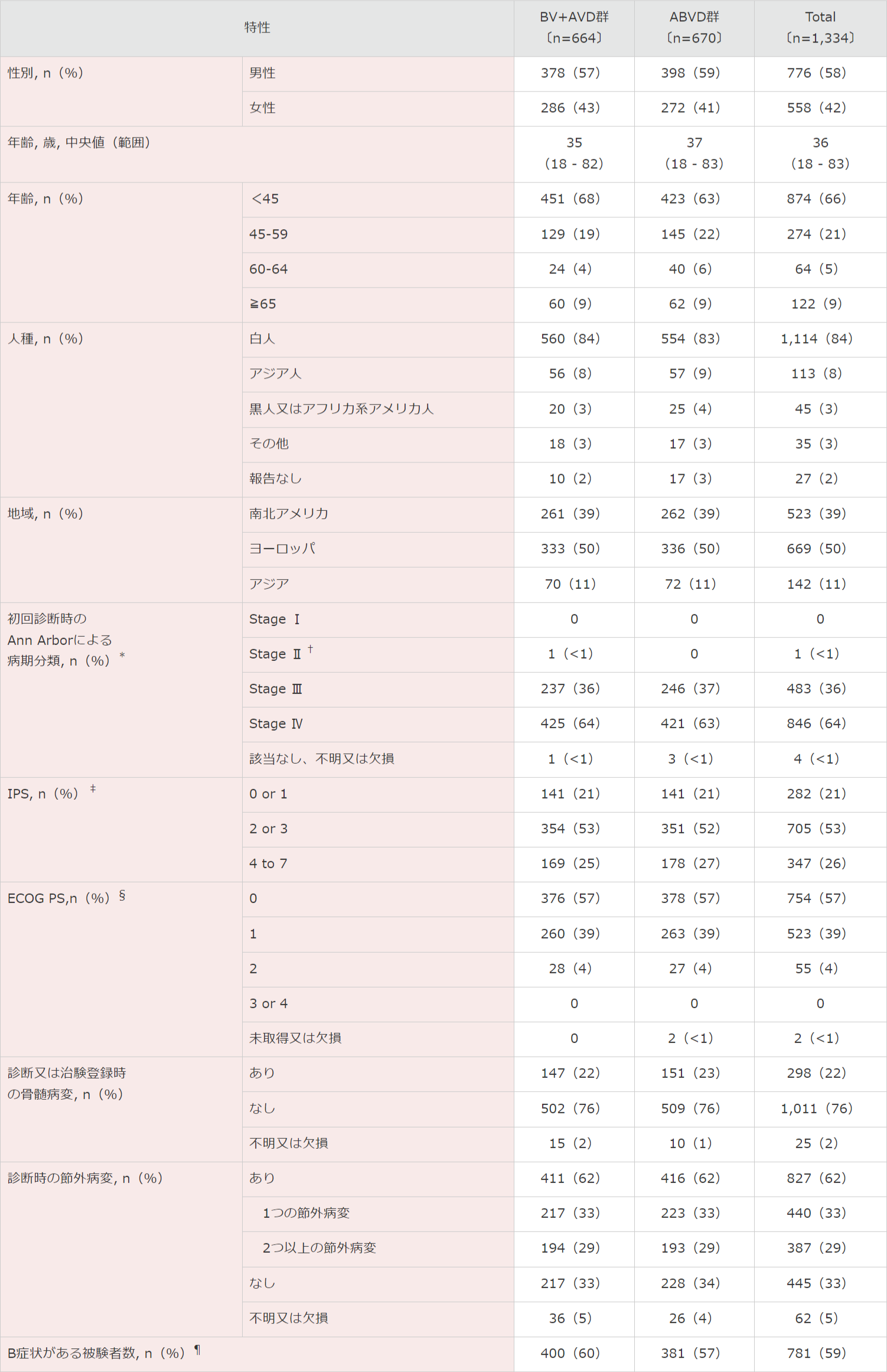
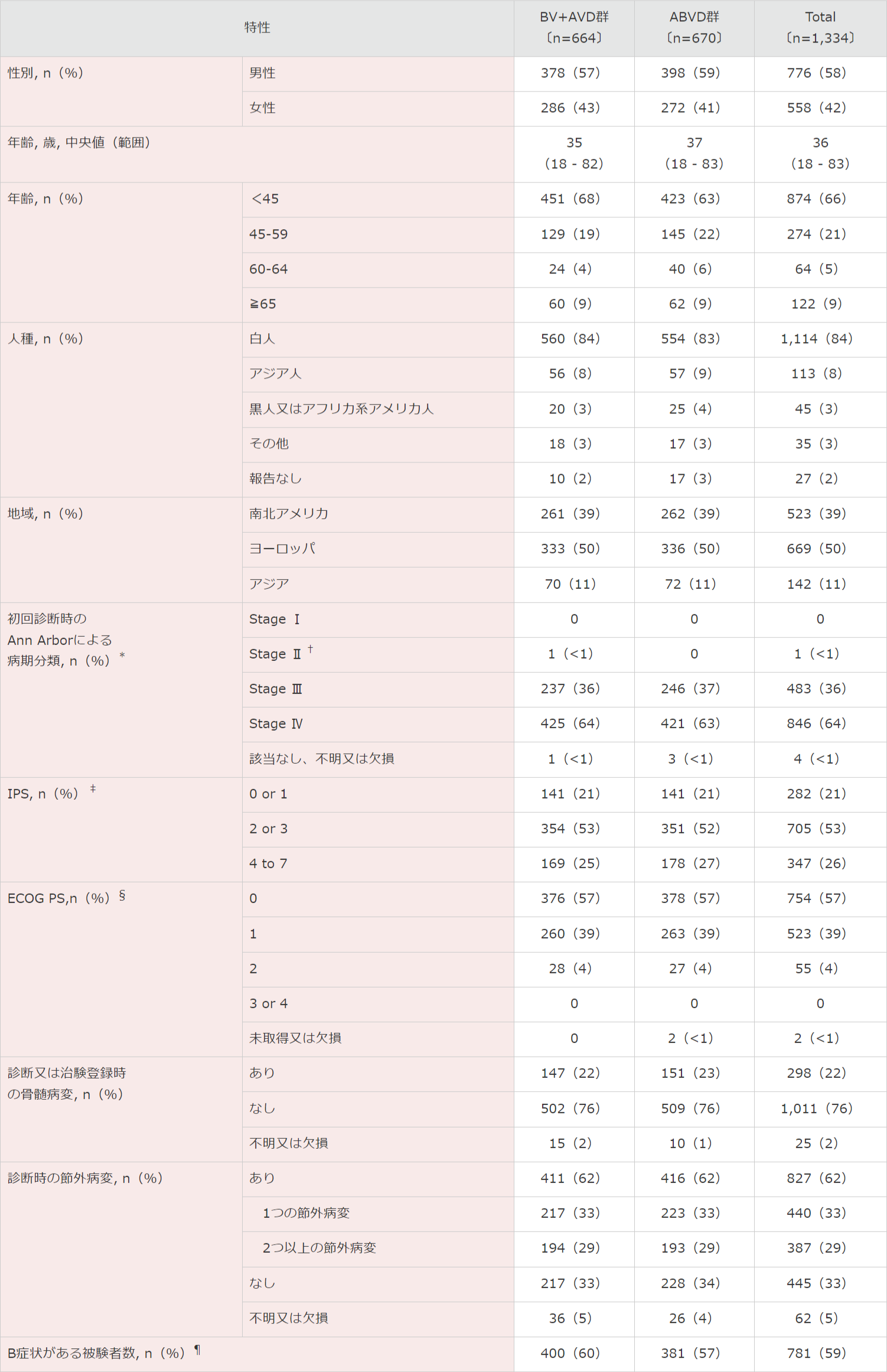
患者背景(ITT解析対象集団)
IPS:国際予後スコア。
ECOG PS:米国東海岸がん臨床試験グループのパフォーマンスステータス。
* Ann Arbor1)による病期分類はⅠ〜Ⅳの範囲にあり、より高い病期がより広範な疾患を示す。
† この区分の患者は、プロトコル違反。
‡ IPS2)は0~7で評価し、高いスコアが治療失敗のリスクの増加を示す。スコア0~1は低リスク、スコア2~3は中等度リスク、スコア4~7は高リスク。
§ ECOG PS3)は0~5で評価し、高いスコアがより大きい身体障害を示す。
¶ B症状は、盗汗、原因不明の発熱(体温>38℃)、又は10%以上の体重減少からなる。
1) Lister TA, et al. : J Clin Oncol. 1989; 7 : 1630-1636.
2) Hasenclever D, et al. : N Engl J Med. 1998; 339 : 1506-1514.
3) Oken MM, et al. : Am J Clin Oncol. 1982; 5 : 649-655.
IPS:国際予後スコア。
ECOG PS:米国東海岸がん臨床試験グループのパフォーマンスステータス。
* Ann Arbor1)による病期分類はⅠ〜Ⅳの範囲にあり、より高い病期がより広範な疾患を示す。
† この区分の患者は、プロトコル違反。
‡ IPS2)は0~7で評価し、高いスコアが治療失敗のリスクの増加を示す。スコア0~1は低リスク、スコア2~3は中等度リスク、スコア4~7は高リスク。
§ ECOG PS3)は0~5で評価し、高いスコアがより大きい身体障害を示す。
¶ B症状は、盗汗、原因不明の発熱(体温>38℃)、又は10%以上の体重減少からなる。
1) Lister TA, et al. : J Clin Oncol. 1989; 7 : 1630-1636.
2) Hasenclever D, et al. : N Engl J Med. 1998; 339 : 1506-1514.
3) Oken MM, et al. : Am J Clin Oncol. 1982; 5 : 649-655.
国際共同第Ⅲ相試験(C25003試験; ECHELON-1試験)(初回解析)
(1)mPFS[主要評価項⽬](検証項⽬)
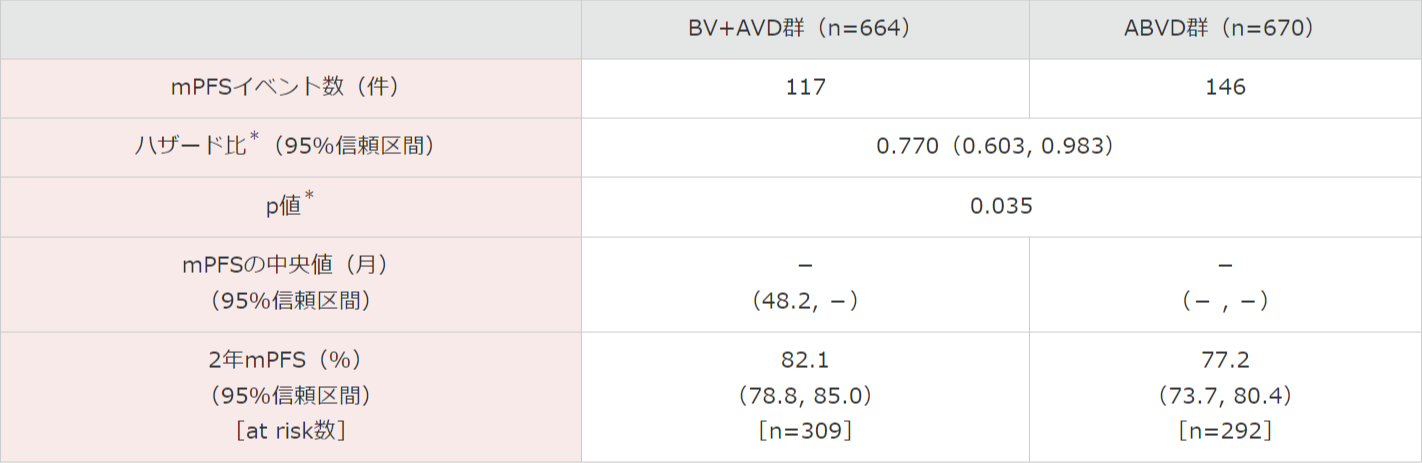
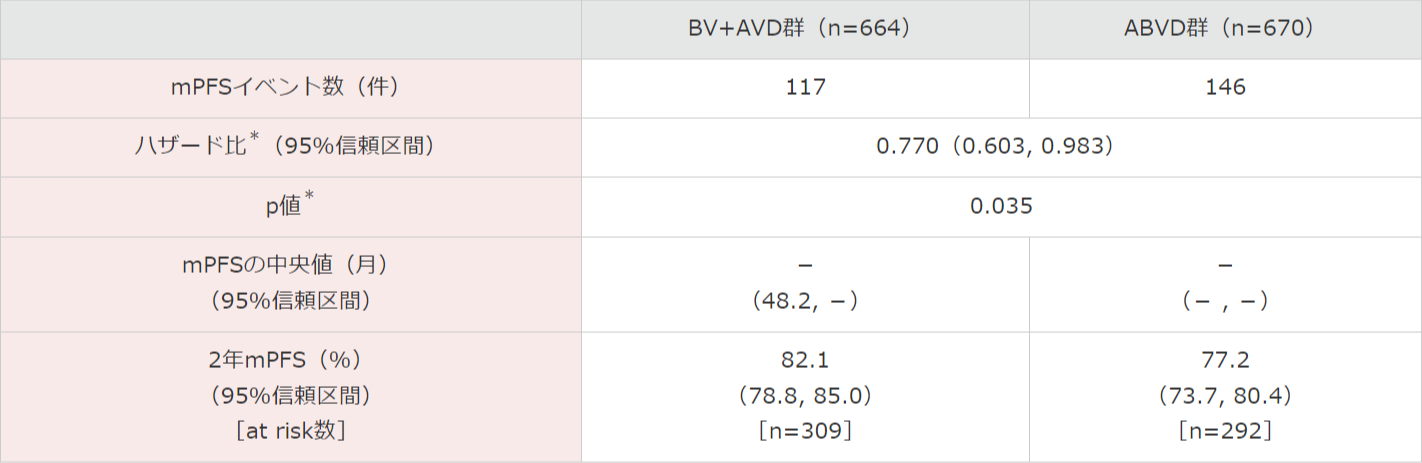
mPFSの中央値は両群とも推定不能でしたが、ランダム化層別因子(地域、ベースライン時のIPSリスク因子)による層別ログランク検定を行った結果、BV+AVD群ではABVD群と比較して統計学的に有意なmPFSの延長が認められました(p=0.035)。
その結果、BV+AVD群のABVD群に対する優越性が検証されました。また、層別*Cox回帰モデルを用いたハザード比は0.770(95%信頼区間:0.603, 0.983)であり、BV+AVD群ではABVD群に比べてmPFSイベントが23.0%軽減しました〔中央判定委員会(以下IRF)判定、追跡調査期間の中央値が両群ともに24.6ヵ月〕。
mPFSイベントが認められなかった被験者は、2年時点でBV+AVD群の82.1%(95%信頼区間:78.8, 85.0)及びABVD群の77.2%(95%信頼区間:73.7, 80.4)と推定されました。
■ mPFSのKaplan-Meier曲線(ITT解析対象集団)(IRF判定)[主要評価項⽬]
初回解析
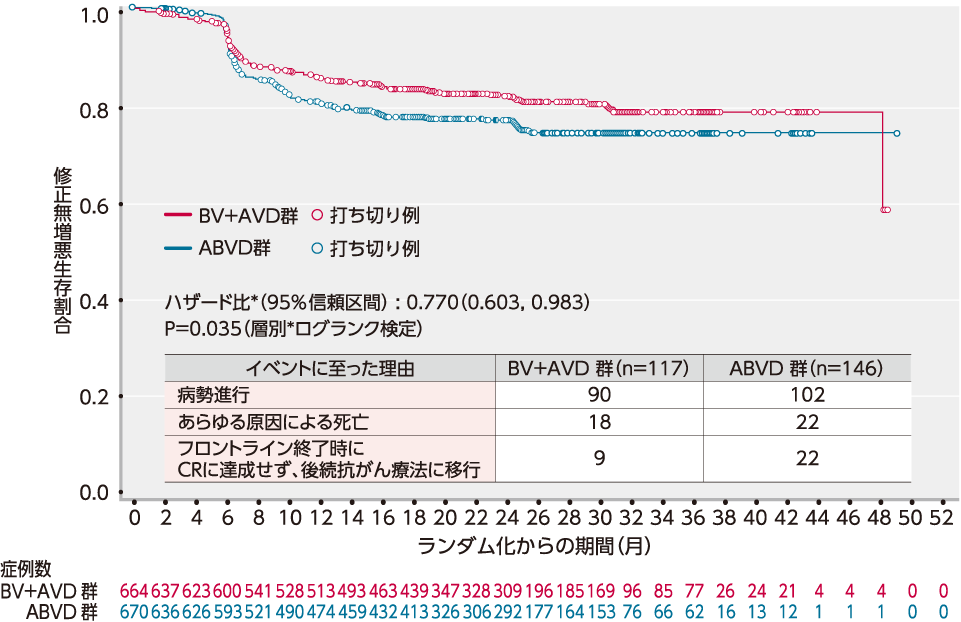
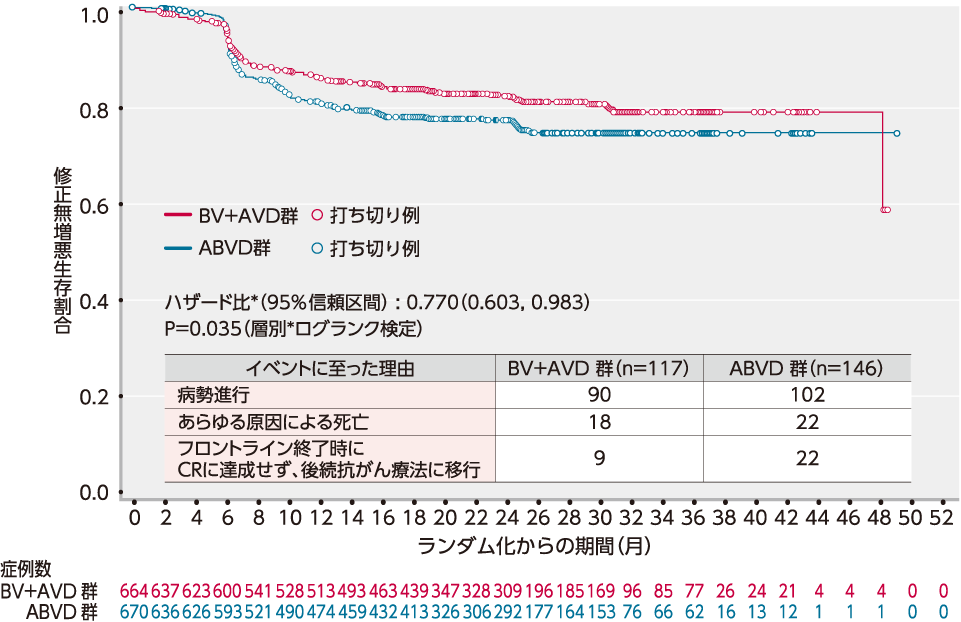
* P値は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別ログランク検定により算出した。またハザード比は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別Cox回帰モデルにより算出した。
* P値は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別ログランク検定により算出した。またハザード比は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別Cox回帰モデルにより算出した。
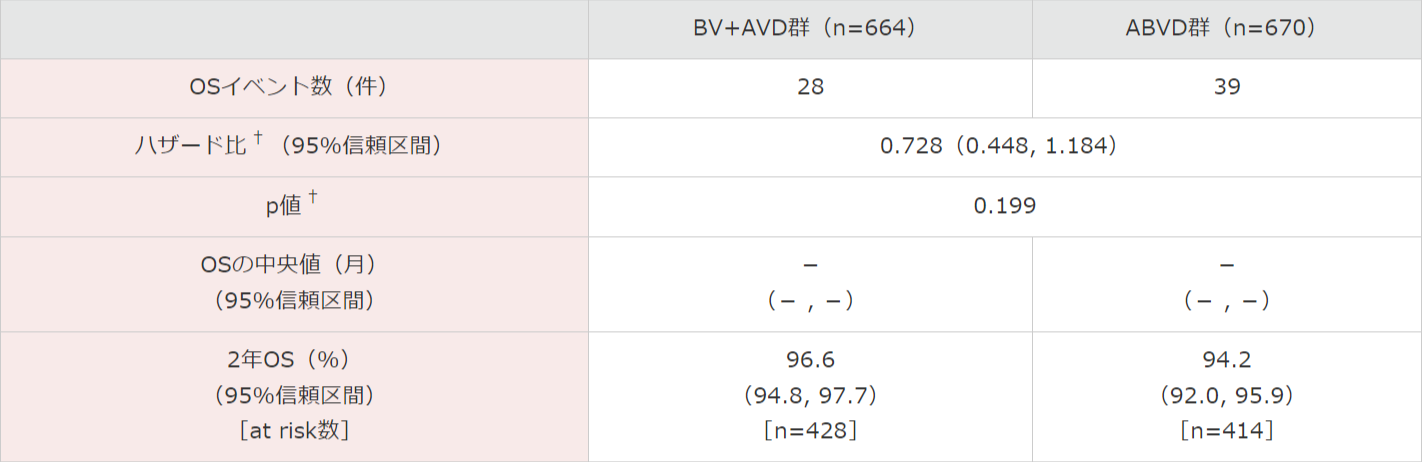
(2)OS[重要な副次評価項目]
OSの初回解析*では、BV+AVD群の28例、ABVD群の39例の死亡が認められました(追跡調査期間の中央値がBV+AVD群で27.8ヵ月、ABVD群で27.4ヵ月の時点)。
OSの中央値は両群とも推定不能でした〔ハザード比:0.728, 95%信頼区間:0.448, 1.184(層別†Cox回帰モデル)、p=0.199(層別†ログランク検定)〕。
OS率は、2年時点でBV+AVD群では96.6%(95%信頼区間:94.8, 97.7)、ABVD群では94.2%(95%信頼区間:92.0, 95.9)と推定されました。
*OSの初回解析はmPFSの最終解析時に実施。2回目の中間解析は当局と相談の元103例のイベントが認められた時点で実施。
最終解析は、死亡が112件見られた時点もしくは最終登録症例ランダム化から10年後の早い時点に実施予定。
■ OSのKaplan-Meier曲線(ITT解析対象集団)[重要な副次評価項⽬]
初回解析
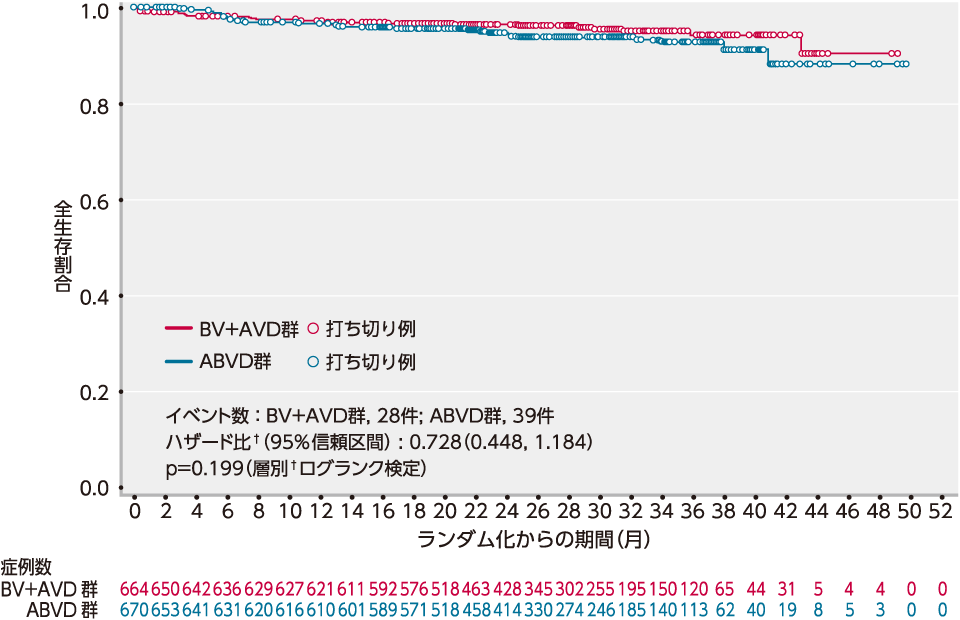
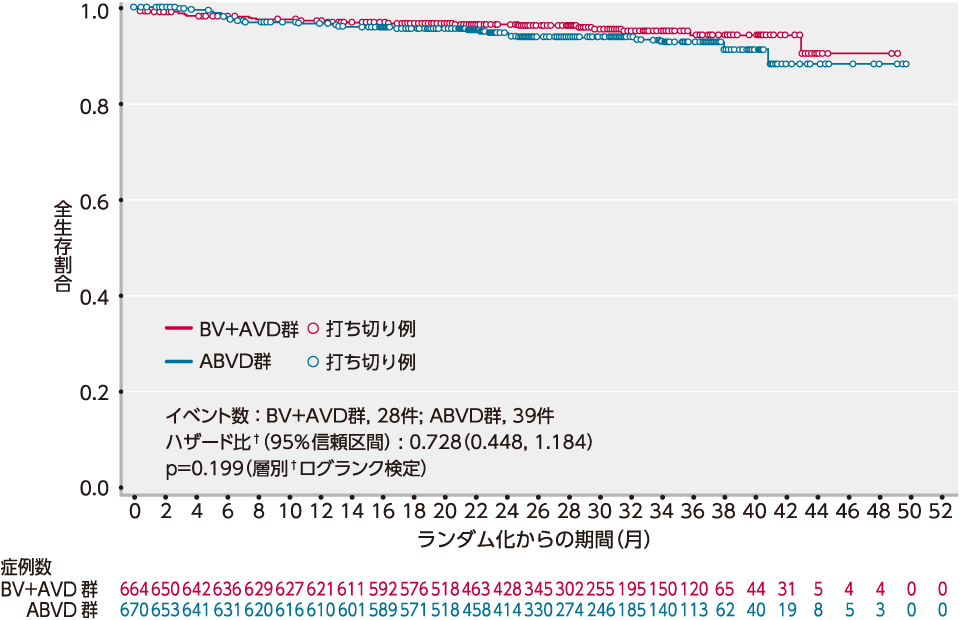
† P値は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別ログランク検定により算出した。またハザード比は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別Cox回帰モデルにより算出した。

† P値は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別ログランク検定により算出した。またハザード比は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別Cox回帰モデルにより算出した。
(3)有効性概要[その他の副次評価項目]
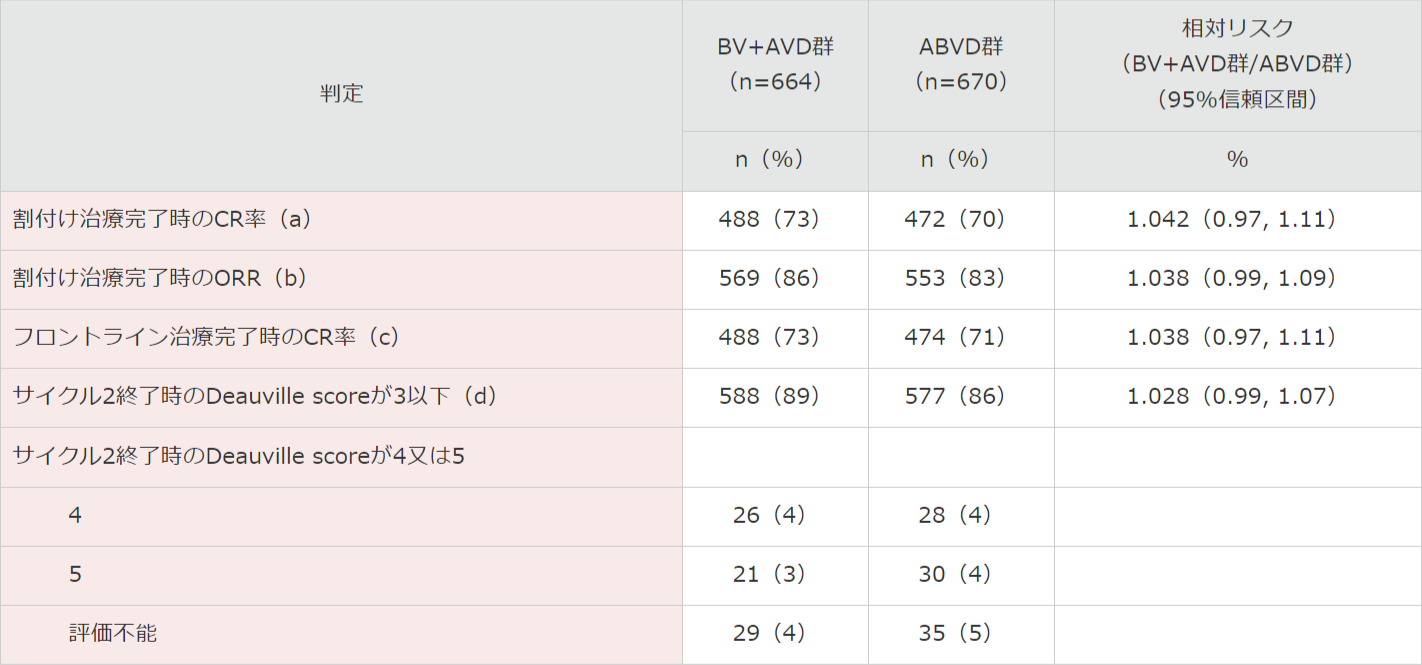
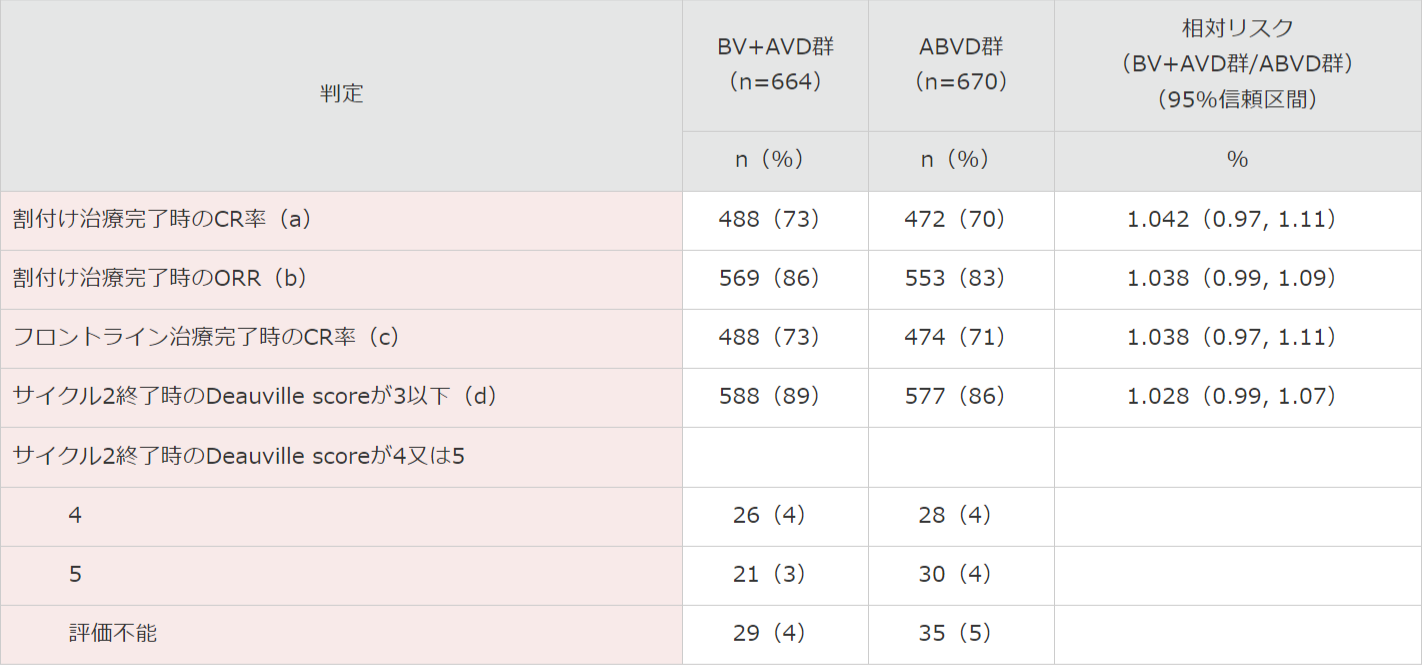
割付け治療完了時(BV+AVD療法もしくはABVD療法を2サイクル終了した時点)のCR率はBV+AVD群で73%、ABVD群で70%(IRF判定)でした†。
割付け治療完了時の全奏効率(ORR)では、BV+AVD群で86%、ABVD群で83%(IRF判定)でした‡。
フロントライン治療完了時(6サイクル終了時点)のCR率は、BV+AVD群で73%、ABVD群で71%(IRF判定)でした†。
サイクル2終了時のPET陰性率(Deauville scoreが3以下)はBV+AVD群で89%、ABVD群で86%でした§。
† 相対リスク及びオッズ比とそれらの95%信頼区間を算出。治療群間におけるCR率の差の95%信頼区間を算出。
‡ BV+AVD又はABVDのいずれかの治療の割付け治療完了時のIRF判定によるCR又はPRした症例の割合を算出。相対リスク及びオッズ比とそれらの95%信頼区間を算出。
§ サイクル2のPETの結果から治療サイクル2でのDeauville scoreが≤3と判断された陰性被験者の割合を算出。相対リスク及びオッズ比とそれらの95%信頼区間を算出。
■ CR率、ORR率、PET陰性率(ITT解析対象集団)
(IRF判定)[その他の副次評価項⽬]
初回解析
(a) 割付け治療完了時の完全寛解は、BV+AVD又はABVDのいずれかの治療の割付け治療完了時のIRF判定による完全寛解した症例の割合で定義される。
(b) 割付け治療完了時の全奏効率は、BV+AVD又はABVDのいずれかの治療の割付け治療完了時のIRF判定による完全寛解又は部分寛解した症例の割合で定義される。
(c) フロントライン治療完了時の完全寛解は、BV+AVD又はABVDのいずれかの割付け又は、代替フロントライン治療の後にIRF判定による完全寛解した症例の割合で定義される。
(d) サイクル2終了時でのPET陰性率は、サイクル2のPETの結果から治療サイクル2でのDeauville scoreが≤3と判断された陰性被験者の割合で定義される。
(a) 割付け治療完了時の完全寛解は、BV+AVD又はABVDのいずれかの治療の割付け治療完了時のIRF判定による完全寛解した症例の割合で定義される。
(b) 割付け治療完了時の全奏効率は、BV+AVD又はABVDのいずれかの治療の割付け治療完了時のIRF判定による完全寛解又は部分寛解した症例の割合で定義される。
(c) フロントライン治療完了時の完全寛解は、BV+AVD又はABVDのいずれかの割付け又は、代替フロントライン治療の後にIRF判定による完全寛解した症例の割合で定義される。
(d) サイクル2終了時でのPET陰性率は、サイクル2のPETの結果から治療サイクル2でのDeauville scoreが≤3と判断された陰性被験者の割合で定義される。
(4)安全性[その他の副次評価項目]
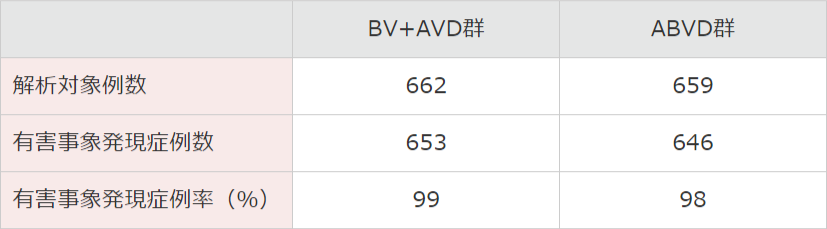
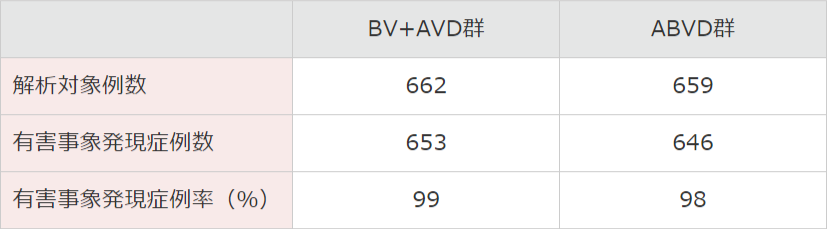
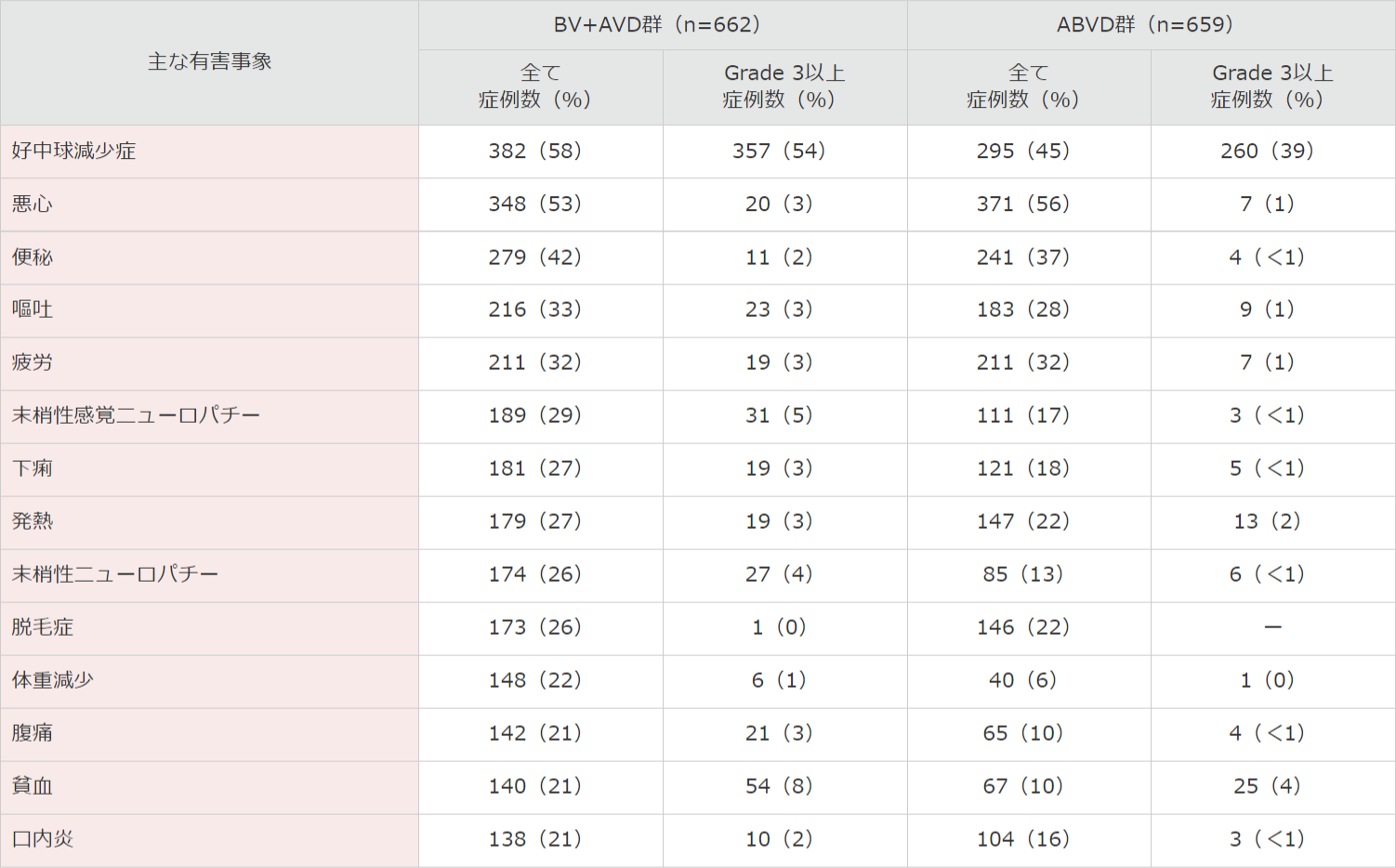
有害事象発現頻度は、BV+AVD群で662例(日本人10例含む)中653例(99%)及びABVD群で659例(日本人13例含む)中646例(98%)でした。主な有害事象(20%以上)は、BV+AVD群では好中球減少症382例(58%)、悪心348例(53%)、便秘279例(42%)、嘔吐216例(33%)、疲労211例(32%)、末梢性感覚ニューロパチー189例(29%)、下痢181例(27%)、発熱179例(27%)、末梢性ニューロパチー174例(26%)、脱毛症173例(26%)、体重減少148例(22%)、腹痛142例(21%)、貧血140例(21%)及び口内炎138例(21%)であり、ABVD群では悪心371例(56%)、好中球減少症295例(45%)、便秘241例(37%)、疲労221例(32%)、嘔吐183例(28%)、発熱147例(22%)及び脱毛症146例(22%)でした。
重篤な有害事象はBV+AVD群で284例(43%)及びABVD群で178例(27%)に認められ、下表のとおりでした。
投与中止に至った有害事象はBV+AVD群で88例(13%)及びABVD群で105例(16%)でした。有害事象の内訳(いずれかの治療群で2例以上)は、BV+AVD群では、末梢性感覚ニューロパチー23例、末梢性ニューロパチー16例、末梢性運動ニューロパチー10例、発熱性好中球減少症9例、敗血症4例、好中球減少症3例、呼吸困難、感覚鈍麻、肺浸潤、心筋梗塞、多発ニューロパチー、斑状皮疹、敗血症性ショック〔以上、各2例〕、肺炎、肺臓炎〔以上、各1例〕でした。ABVD群では、呼吸困難25例、咳嗽、肺毒性〔以上、各12例〕、一酸化炭素拡散能減少10例、肺臓炎9例、末梢性感覚ニューロパチー6例、発熱性好中球減少症4例、末梢性ニューロパチー、間質性肺疾患、発熱〔以上、各3例〕、肺炎、労作性呼吸困難、呼吸時疼痛〔以上、各2例〕及び末梢性運動ニューロパチー、好中球減少症、心筋梗塞、多発ニューロパチー〔以上、各1例〕でした。
治験期間中の死亡例(治験治療薬の最終投与後30日以内に認められた死亡)はBV+AVD群で9例(1%)及びABVD群で13例(2%)でした。治験期間中の死因の内訳は、BV+AVD群では、心筋梗塞が2例、心肺停止、貪食細胞性組織球症、呼吸不全、(原因不明の)死亡、多臓器機能不全症候群、好中球減少性敗血症及び敗血症性ショックが各1例でした。ABVD群では、肺炎3例、心停止2例、ニューモシスチス・イロベチイ肺炎、肺毒性、心肺不全、肺臓炎、急性呼吸窮迫症候群、呼吸障害、脳血管発作及び原因不明の死亡が各1例でした。死亡が認められたBV+AVD群9例のうち8例及びABVD群13例のうち7例では治験治療薬との因果関係が否定されませんでした。
有害事象発現状況一覧
■ 有害事象の発現状況
初回解析
■ 有害事象の種類別発現頻度
国際共同第Ⅲ相試験(C25003試験)においていずれかの群で20%以上認められた有害事象
<併用投与>(安全性解析対象集団)
初回解析
MedDRA Ver.19.0、NCI-CTCAE Ver.4.03により集計。国際共同第Ⅲ相試験(C25003試験)。
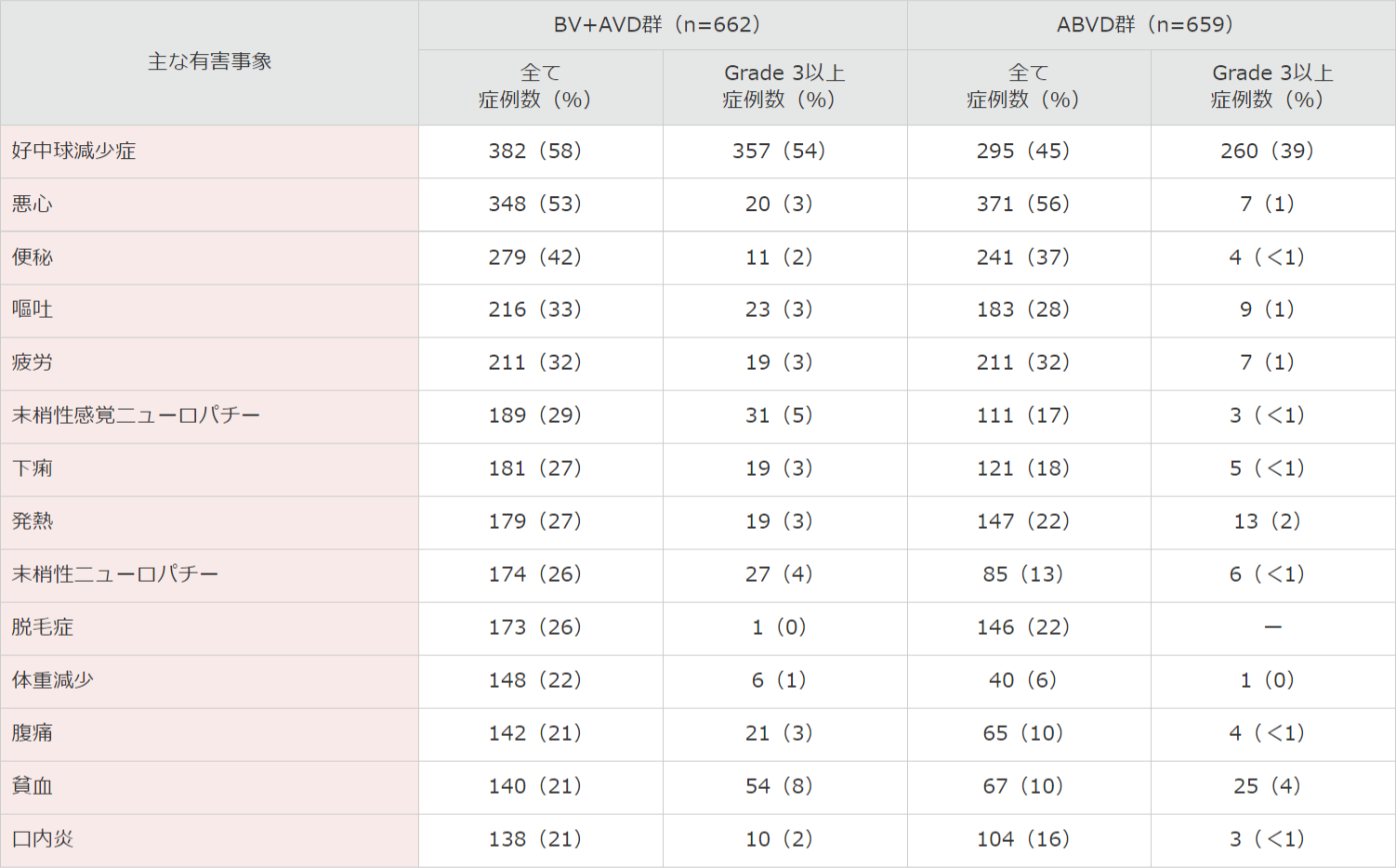
MedDRA Ver.19.0、NCI-CTCAE Ver.4.03により集計。国際共同第Ⅲ相試験(C25003試験)。
■ 国際共同第Ⅲ相試験(C25003試験)における重篤な有害事象*<併用投与>(安全性解析対象集団)
初回解析
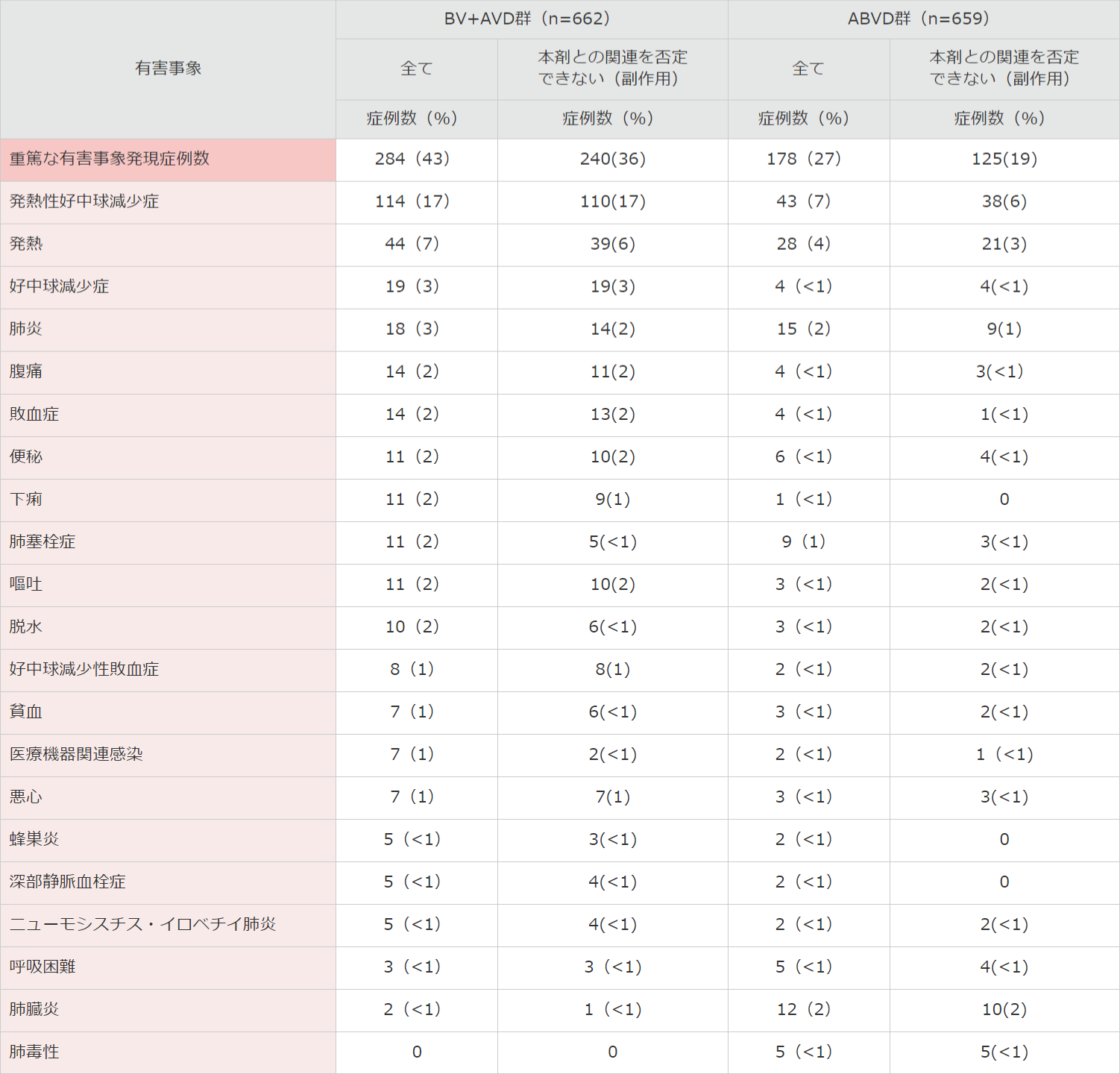
MedDRA Ver.19.0、NCI-CTCAE Ver.4.03により集計。
C25003試験。
*いずれかの治療群で5名以上発現した重篤な有害事象を示した。
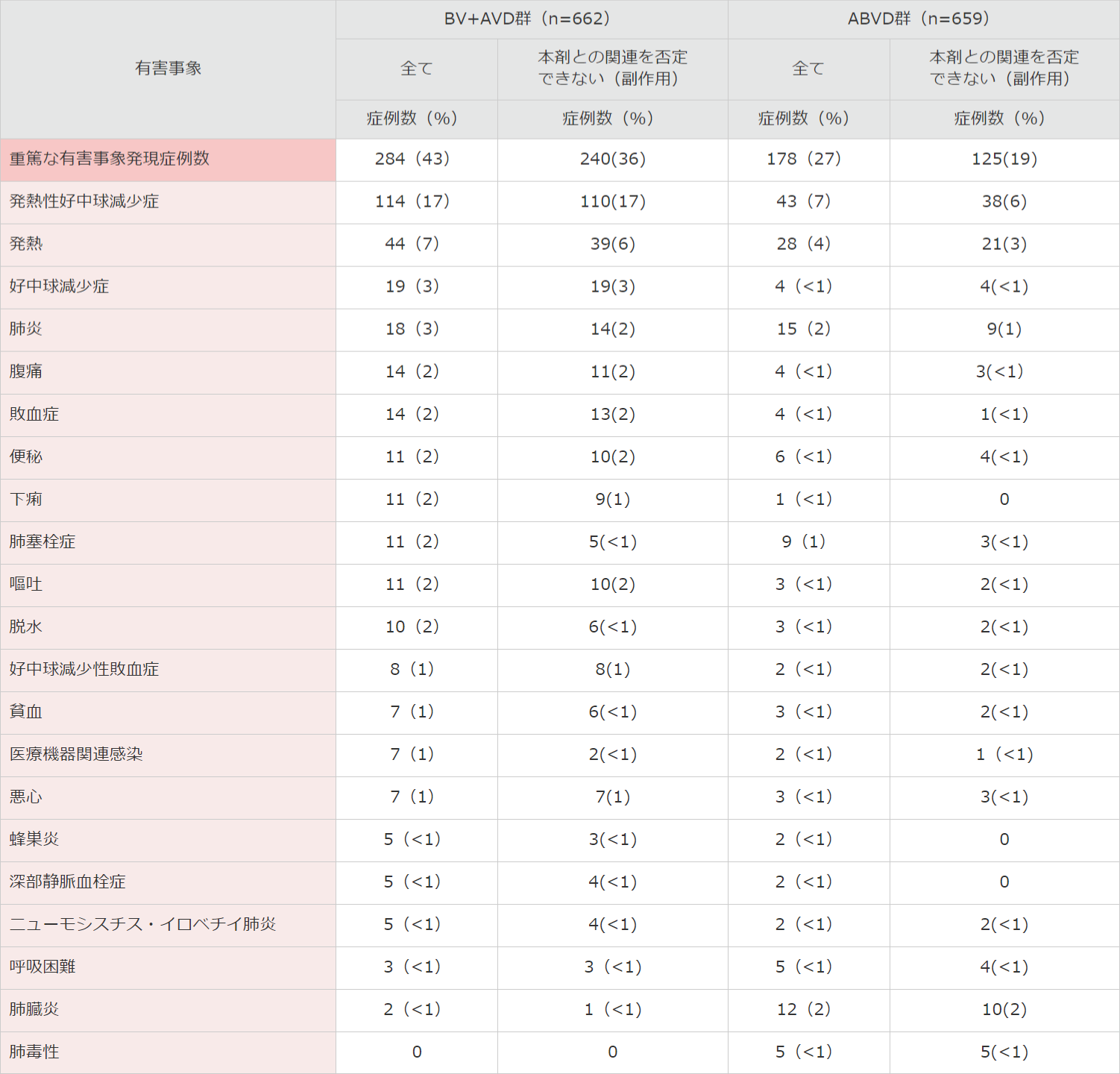
MedDRA Ver.19.0、NCI-CTCAE Ver.4.03により集計。
C25003試験。
*いずれかの治療群で5名以上発現した重篤な有害事象を示した。
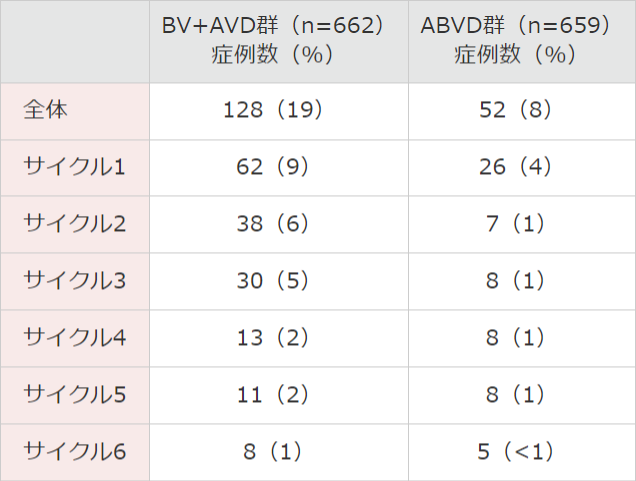
■ 発熱性好中球減少症(FN)の発現状況注)(安全性解析対象集団)
FNは両治療群ともにサイクル1で最も多く認められました。サイクル1で認められたFNは、BV+AVD群で62例(9%)、ABVD群で26例(4%)でした。
● 投与サイクルごとのFN
初回解析
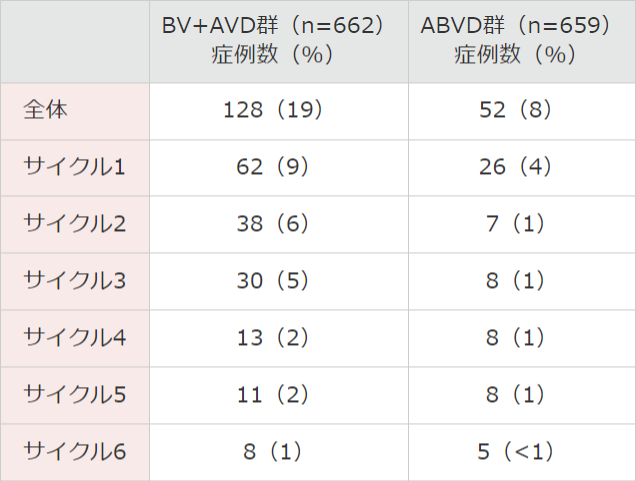
%:安全性解析対象集団の被験者数に基づく。
被験者ごとのCTCAEの最大Gradeを要約。
最も高いGradeで被験者を1回カウント。
%:安全性解析対象集団の被験者数に基づく。
被験者ごとのCTCAEの最大Gradeを要約。
最も高いGradeで被験者を1回カウント。
● G-CSFの予防的投与*別の有害事象[サブグループ解析]
● G-CSFの予防的投与*別の有害事象[サブグループ解析]
BV+AVD群では、G-CSF製剤を用いた一次予防*を受けた83例のFNの発症率は11%(83例中9例)、予防投与を受けなかった被験者は21%(579例中119例)に認められました。
Grade 3以上の感染症及び寄生虫症はG-CSFの予防的投与を受けた被験者では11%(83例中9例)、予防的投与を受けなかった被験者は18%(579例中107例)に認められました**。
*治験実施計画書では、BV+AVD群の被験者の好中球減少症の管理のために、実施医療機関の使用指針に基づくG-CSFの使用を許容した。予定する被験者数の約70%の登録後、BV+AVD群に割り付けられた被験者に対し、サイクル1の開始時にG-CSFの予防的投与を推奨することを治験責任医師に連絡した。
**治験薬投薬投与日(1日目)から5日目までにG-CSFを投与した場合と定義した。重要な有害事象に関連するリスク因子(G-CSFの予防的投与の有無、年齢60歳以上)に関して、G-CSFの予防的投与の有無別の発熱性好中球減少症及び関連するその他の有害事象の発現頻度、60歳以上の被験者における有害事象のサブグループ解析を事後解析として実施した。解析結果は、承認審査評価資料に記載し、評価された。
初回解析
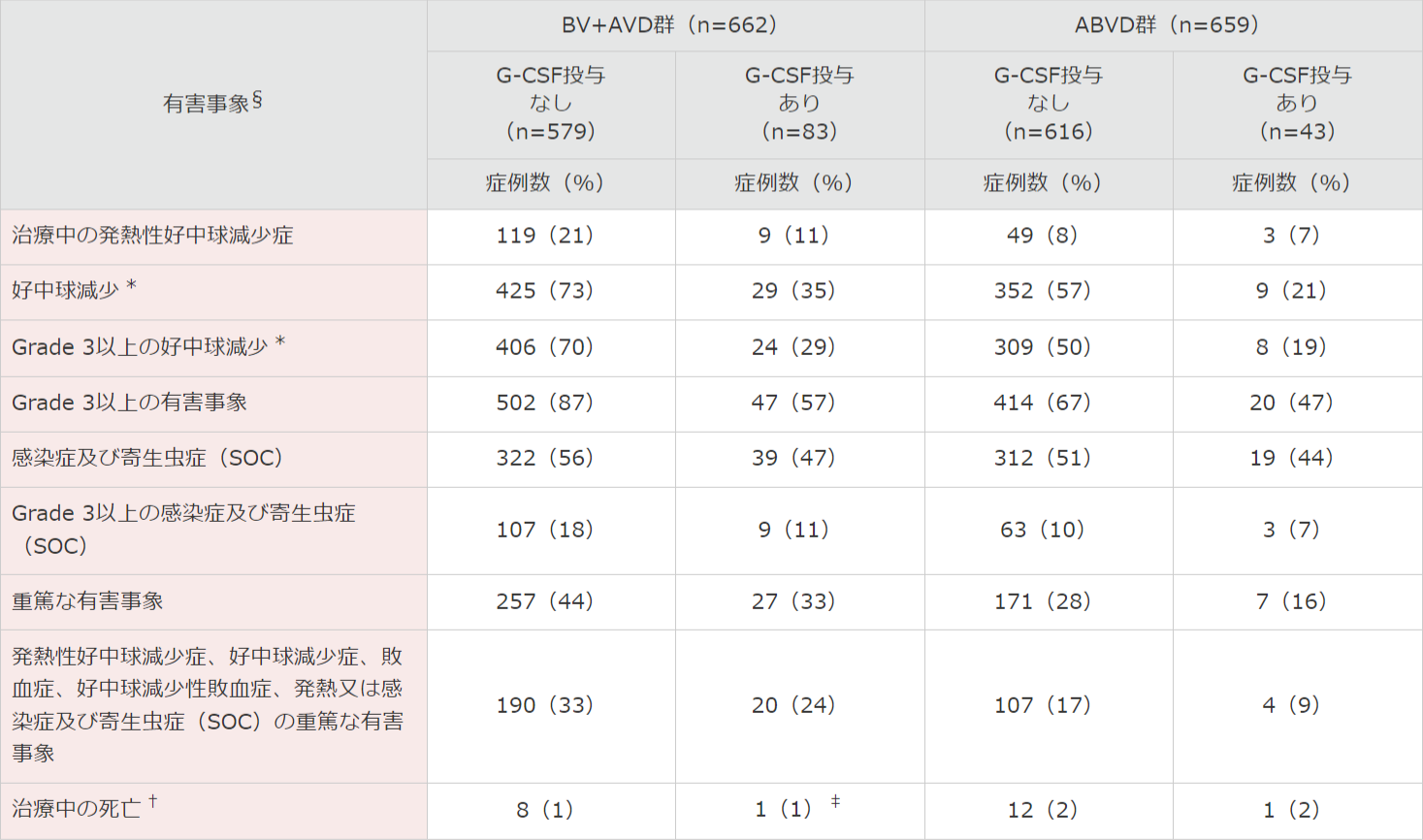
§ 本剤との関連が否定された事象を含む。
* 好中球減少およびGrade 3以上の好中球減少(好中球数<1,000個/mm3)には、“好中球減少症”と“好中球数減少”を含む。
† 治験治療薬の最終投与後30日以内に認められた死亡と定義。
‡ 薬剤投与5日目より前(G-CSF製剤一次予防的投与前)に発症した好中球減少症の治療のためにG-CSF製剤投与を受けた。
SOC:MedDRAの器官別大分類。
MedDRA Ver.19.0、 NCI-CTCAE Ver.4.03により集計。
G-CSF製剤の使用にあたっては各社の電子添文をご確認ください。
注)その他の副次評価項目である安全性に関して、ブレンツキシマブ ベドチンの医薬品リスク管理計画の【重要な特定されたリスク】の一つである骨髄抑制のうち発熱性好中球減少症を取り上げました。
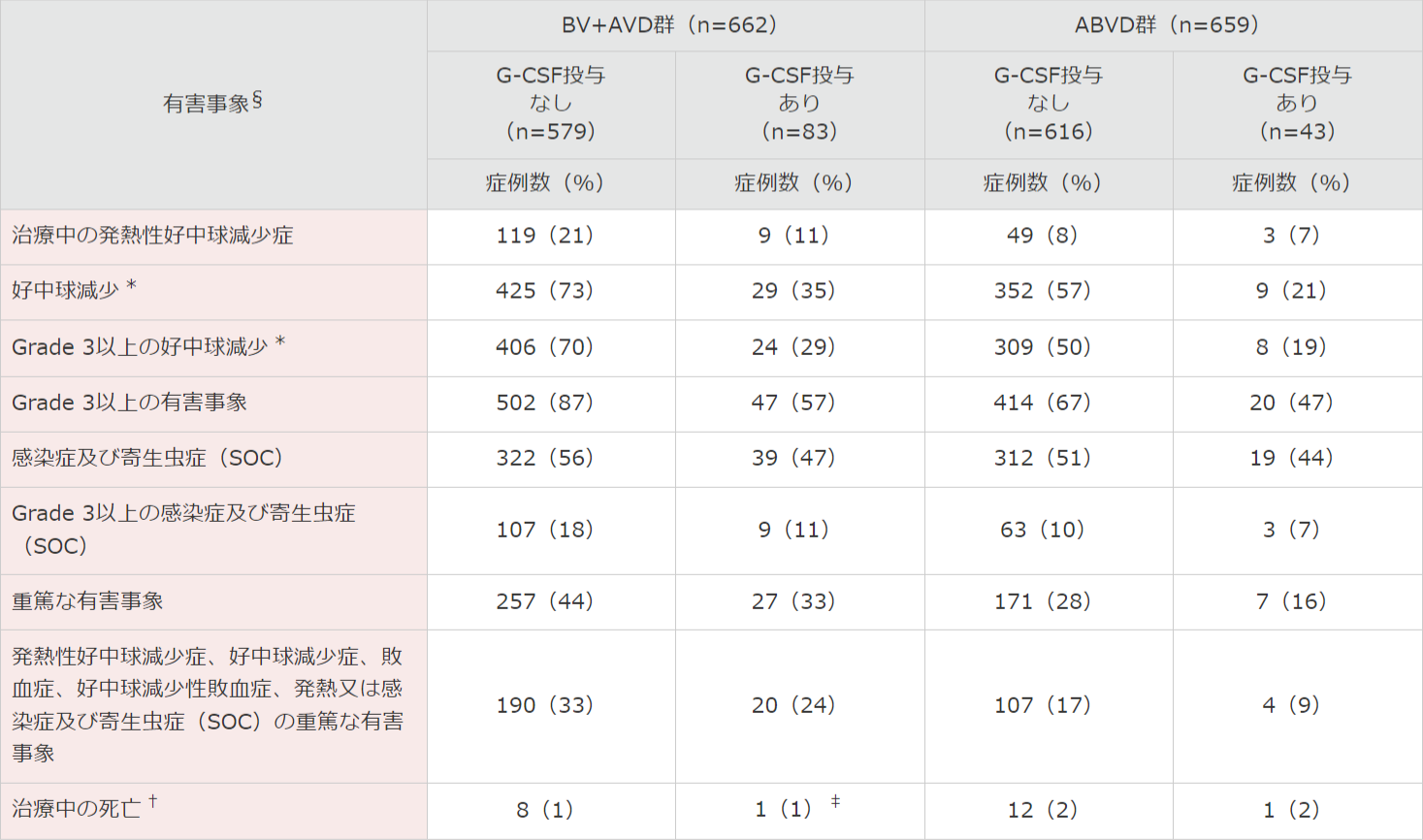
§ 本剤との関連が否定された事象を含む。
* 好中球減少およびGrade 3以上の好中球減少(好中球数<1,000個/mm3)には、“好中球減少症”と“好中球数減少”を含む。
† 治験治療薬の最終投与後30日以内に認められた死亡と定義。
‡ 薬剤投与5日目より前(G-CSF製剤一次予防的投与前)に発症した好中球減少症の治療のためにG-CSF製剤投与を受けた。
SOC:MedDRAの器官別大分類。
MedDRA Ver.19.0、 NCI-CTCAE Ver.4.03により集計。
G-CSF製剤の使用にあたっては各社の電子添文をご確認ください。
注)その他の副次評価項目である安全性に関して、ブレンツキシマブ ベドチンの医薬品リスク管理計画の【重要な特定されたリスク】の一つである骨髄抑制のうち発熱性好中球減少症を取り上げました。
長期成績を確認する
国際共同第Ⅲ相試験(C25003試験; ECHELON-1試験)の
治験責任医師判定による追跡調査結果・約6年(2回目の中間解析)*
Ansell SM, et al.: N Engl J Med. 2022; 387(4): 310-320.
Ansell SM, et al.: N Engl J Med. 2022; 387(4): 310-320. Supplementary appendix.
本試験は、Takeda Development Center Americas社とSeagen社の資金提供により実施された。
本論文の著者のうちそれぞれ2名は同社の社員で、試験計画、解析、執筆等の支援を受けている。
著者に同社より研究支援、謝礼金等を受領している者が含まれる。
OS[重要な副次評価項目]
OSの2回目の中間解析*では、BV+AVD群が39例、ABVD群が64例の死亡が認められました〔追跡期間中央値は73.0ヵ月(95%信頼区間:72.3-73.6、範囲:0.0-100.6)〕。
治験責任医師判定によるOSは、層別†Cox回帰モデルを用いたハザード比は0.59(95%信頼区間:0.40-0.88)、p=0.009(層別†ログランク検定)と有意に改善し、ブレンツキシマブ ベドチンの投与は死亡のリスクを41%低下させました。
また、6年全生存率推定値は、BV+AVD群が93.9%(95%信頼区間:91.6-95.5)、ABVD群が89.4%(95%信頼区間:86.6-91.7)でした。
*OSの2回目の中間解析は当局と相談の元103例のイベントが認められた時点で実施。最終解析は、死亡が112件見られた時点もしくは最終登録症例ランダム化から10年後の早い時点に実施予定。
■ OSのKaplan-Meier曲線(ITT解析対象集団)[重要な副次評価項目]
2回目の中間解析
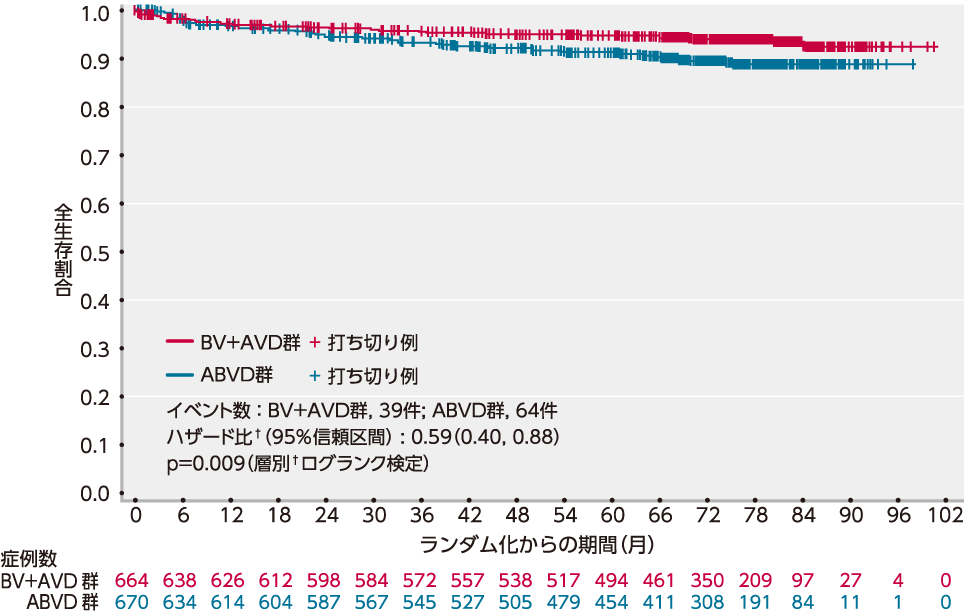
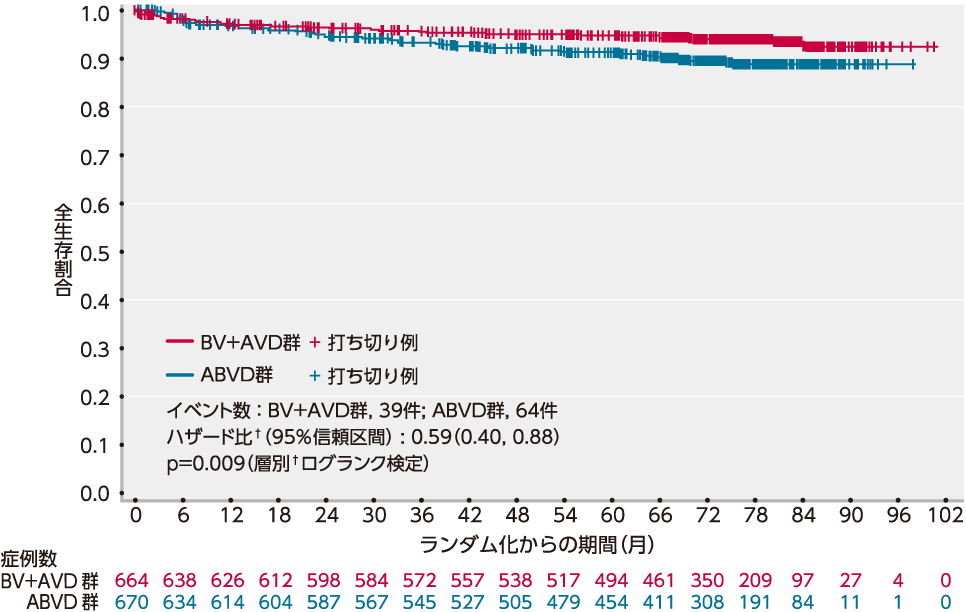

† P値は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別ログランク検定により算出した。またハザード比は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別Cox回帰モデルにより算出した。
重要な副次評価項目のサブグループ解析である治験責任医師判定によるベースライン特性、予後因子別のOSの結果は、60歳未満の被験者、病期分類Ⅳの被験者、IPS4-7の被験者、北アメリカの被験者においてBV+AVD群のハザード比の点推定値はそれぞれ0.51、0.48、0.48、0.33でした。

† P値は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別ログランク検定により算出した。またハザード比は、層別因子(地域、ベースライン時のIPSリスク因子)を用いた層別Cox回帰モデルにより算出した。
重要な副次評価項目のサブグループ解析である治験責任医師判定によるベースライン特性、予後因子別のOSの結果は、60歳未満の被験者、病期分類Ⅳの被験者、IPS4-7の被験者、北アメリカの被験者においてBV+AVD群のハザード比の点推定値はそれぞれ0.51、0.48、0.48、0.33でした。
■ ベースライン特性、予後因子別のOSのフォレストプロット(ITT解析対象集団)
[重要な副次評価項目][サブグループ解析]
2回目の中間解析
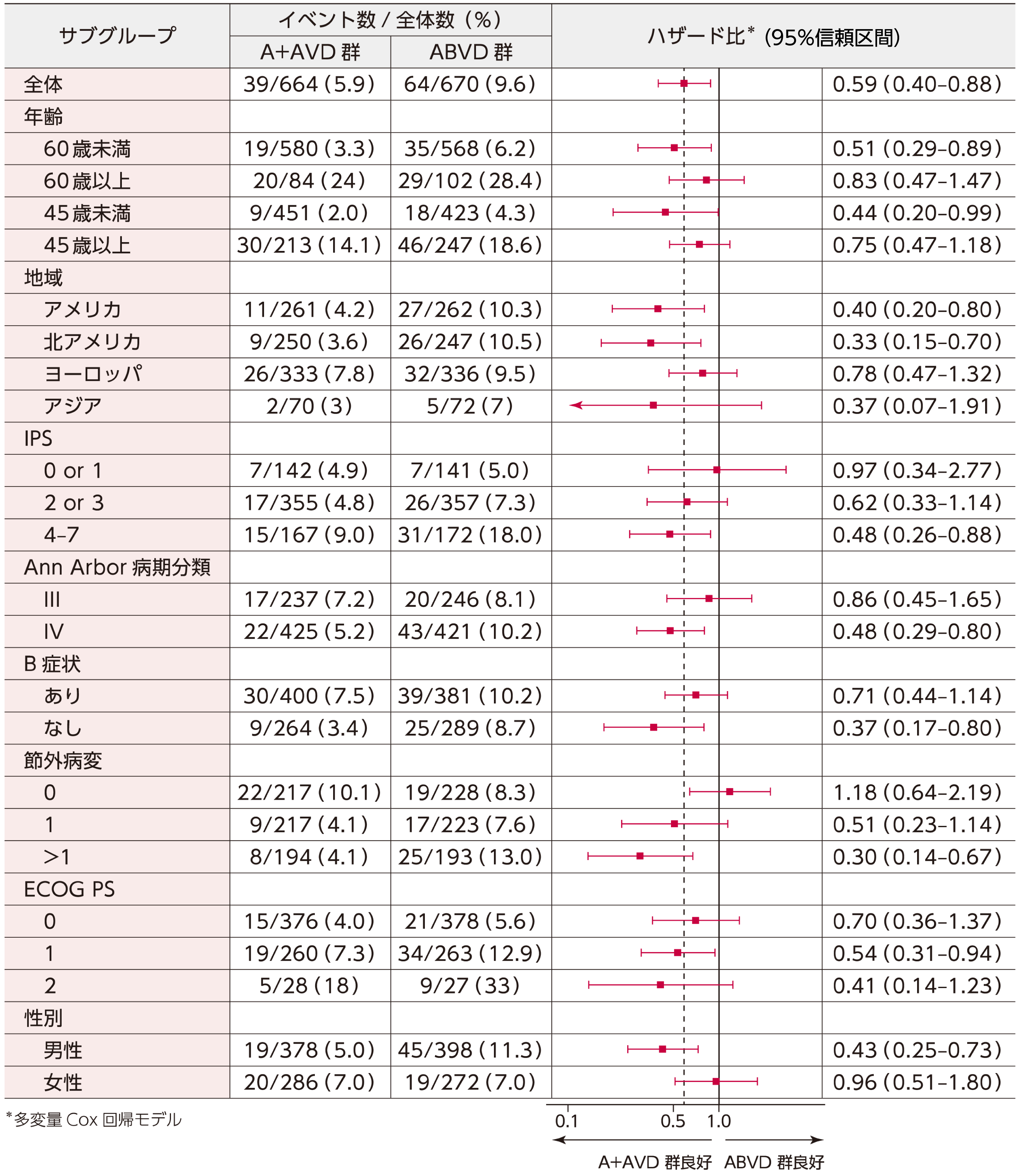
フォレストプロットは、事前に指定したサブグループを用いて治験責任医師判定で行った。多変量Cox回帰モデルを用いて、ABVD群に対するBV+AVD群のハザード比及び95%信頼区間を算出した。ITT Populationは、ランダム化されたすべての症例を含む。IPSは0~7で評価し、高いスコアが治療失敗のリスクの増加を示す。スコア0~1は低リスク、スコア2~3は中等度リスク、スコア4~7は高リスク。B症状は、体重減少、寝汗、発熱と定義した。ECOGパフォーマンスステータスのスコアは5段階で評価し、スコアが高いほど障害があることを示す。破線の縦線は、全試験集団における治療効果を示す。アジアサブグループのハザード比の矢印は、95%信頼区間がグラフの範囲外まで広がっていることを示す。
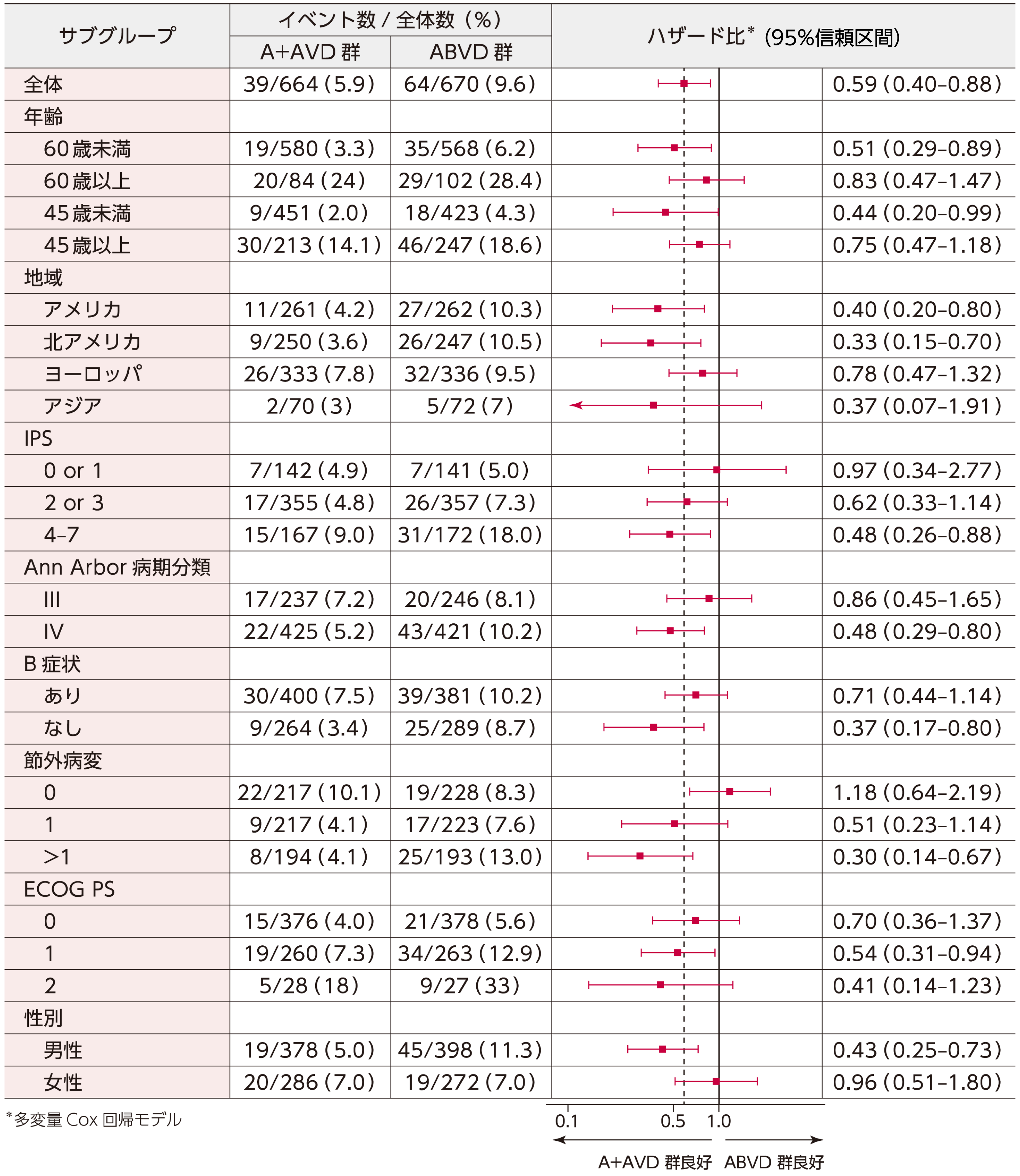
フォレストプロットは、事前に指定したサブグループを用いて治験責任医師判定で行った。多変量Cox回帰モデルを用いて、ABVD群に対するBV+AVD群のハザード比及び95%信頼区間を算出した。ITT Populationは、ランダム化されたすべての症例を含む。IPSは0~7で評価し、高いスコアが治療失敗のリスクの増加を示す。スコア0~1は低リスク、スコア2~3は中等度リスク、スコア4~7は高リスク。B症状は、体重減少、寝汗、発熱と定義した。ECOGパフォーマンスステータスのスコアは5段階で評価し、スコアが高いほど障害があることを示す。破線の縦線は、全試験集団における治療効果を示す。アジアサブグループのハザード比の矢印は、95%信頼区間がグラフの範囲外まで広がっていることを示す。
安全性[その他の副次評価項目]
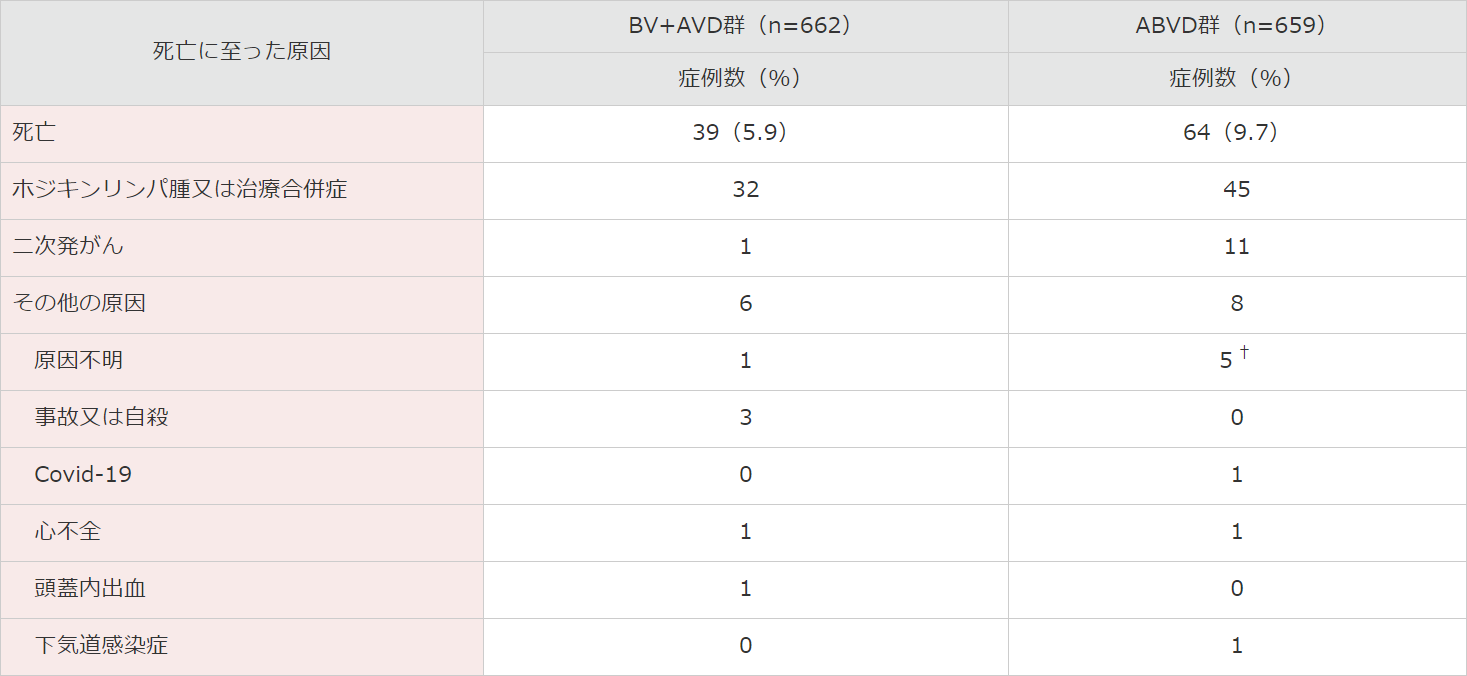
■ 追跡期間中の死因の概要(安全性解析対象集団)
追跡期間中の死因は、BV+AVD群では死亡39例中32例がホジキンリンパ腫⼜は治療合併症で、⼆次発がんによる死亡は1例でした。ABVD群では死亡64例中45例がホジキンリンパ腫⼜は治療合併症で、⼆次発がんによる死亡は11例でした。
2回目の中間解析
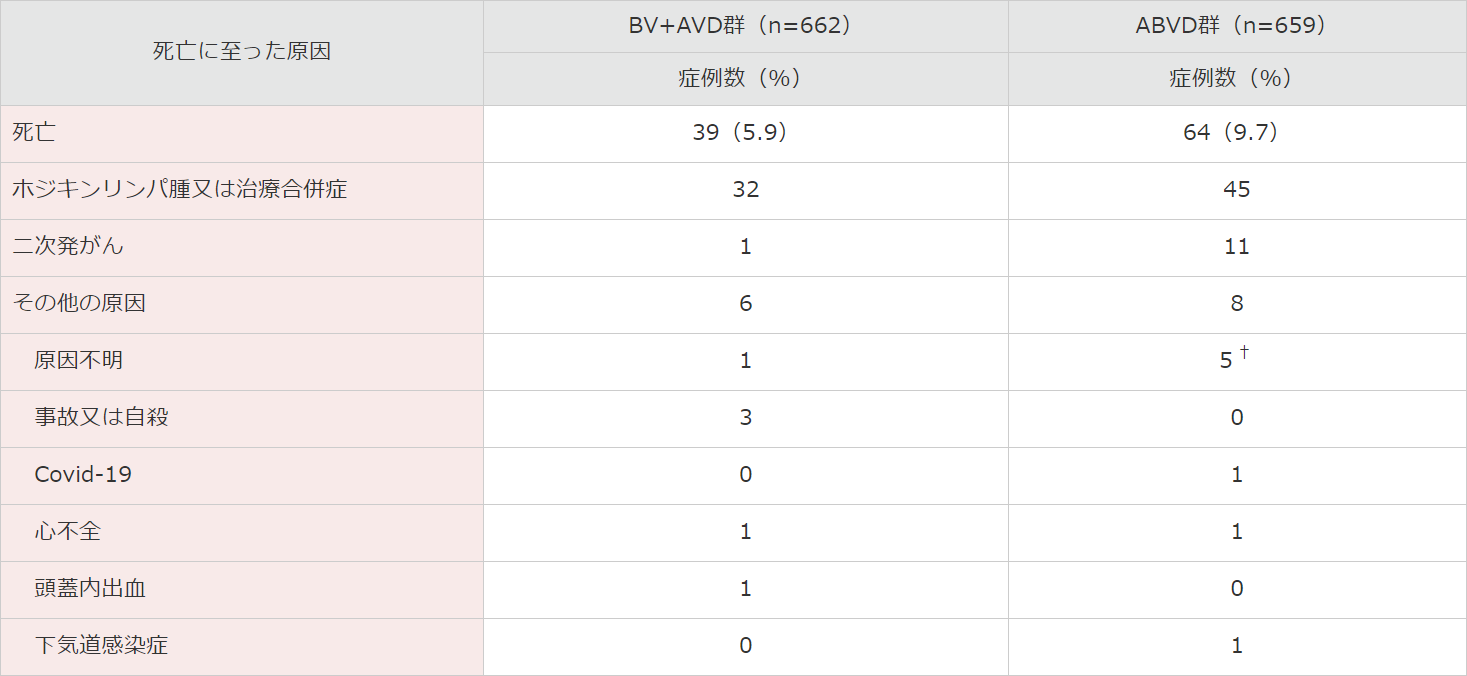
Covid-19:コロナウイルス病2019。
安全性集団には、少なくとも1回の治験薬投与を受けた全患者が含まれる。
† ABVD群2例において、原因不明の死亡が報告されたが、死亡は病勢進⾏後(治験責任医師により記録されたもの)であった。
Covid-19:コロナウイルス病2019。
安全性集団には、少なくとも1回の治験薬投与を受けた全患者が含まれる。
† ABVD群2例において、原因不明の死亡が報告されたが、死亡は病勢進⾏後(治験責任医師により記録されたもの)であった。
■ 治療群別の⼆次発がん(安全性解析対象集団)
二次発がんは55例の被験者で報告され、その内訳はBV+AVD群23例(3.5%)、ABVD群32例(4.9%)でした。BV+AVD群では固形腫瘍が14例、血液学的悪性腫瘍が9例でした。ABVD群では固形腫瘍が14例、血液学的悪性腫瘍が17例、不明な悪性腫瘍が1例でした。
二次発がんの42%は、60歳以上の被験者に発生しました(BV+AVD群23例中9例[39%]、ABVD群32例中14例[44%])。
2回目の中間解析

* 数字は症例数である。固形腫瘍の内3つ(基底細胞癌、結腸腫瘍、黒色腫)は1例の症例について報告された。

* 数字は症例数である。固形腫瘍の内3つ(基底細胞癌、結腸腫瘍、黒色腫)は1例の症例について報告された。
■ 治療後の妊娠と出産[探索的評価項⽬](安全性解析対象集団)
妊娠は、BV+AVD群において女性被験者は49例、男性被験者のパートナーは33例で報告され、ABVD群において女性被験者は28例、男性被験者のパートナーは33例で報告されました。
また、治験参加者及びそのパートナーにおいて195件の妊娠数が報告されました。その内訳は、BV+AVD群において女性被験者は70件、男性被験者のパートナーは44件で報告され、ABVD群において女性被験者は35件、男性被験者のパートナーは46件で報告されました。
2回目の中間解析
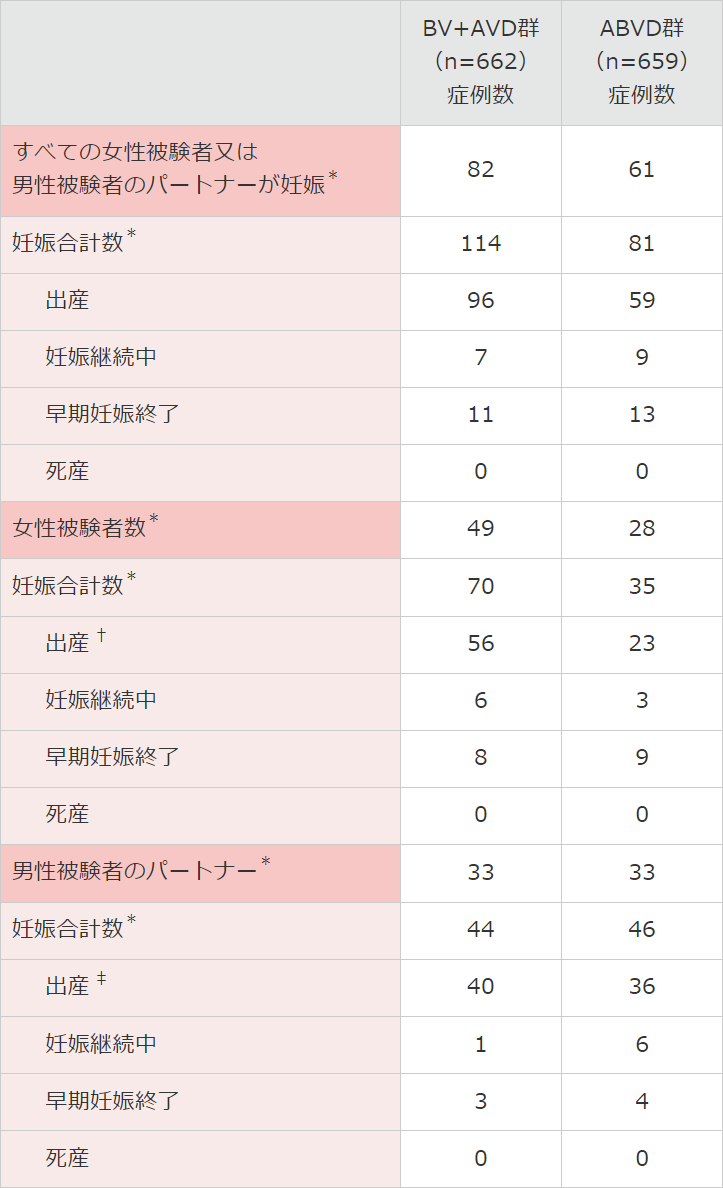
* 1例の女性被験者又は男性被験者のパートナーに関して、複数の妊娠又は妊娠転帰が報告されている場合がある。
† BV+AVD群の13例、及びABVD群の3例の女性被験者について、2回以上の生児出生が記録されている。
‡ BV+AVD群の男性被験者の8例のパートナー及びABVD群の男性被験者の7例のパートナーについて、2回以上の生児出生が記録されている。
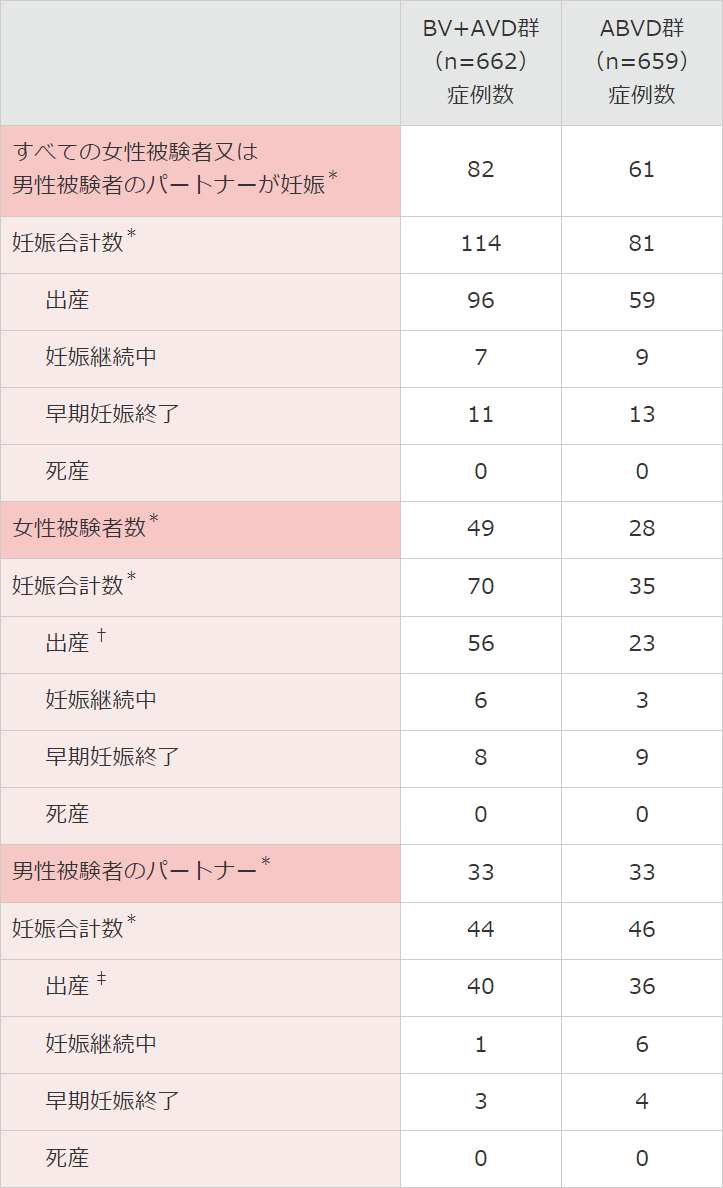
* 1例の女性被験者又は男性被験者のパートナーに関して、複数の妊娠又は妊娠転帰が報告されている場合がある。
† BV+AVD群の13例、及びABVD群の3例の女性被験者について、2回以上の生児出生が記録されている。
‡ BV+AVD群の男性被験者の8例のパートナー及びABVD群の男性被験者の7例のパートナーについて、2回以上の生児出生が記録されている。
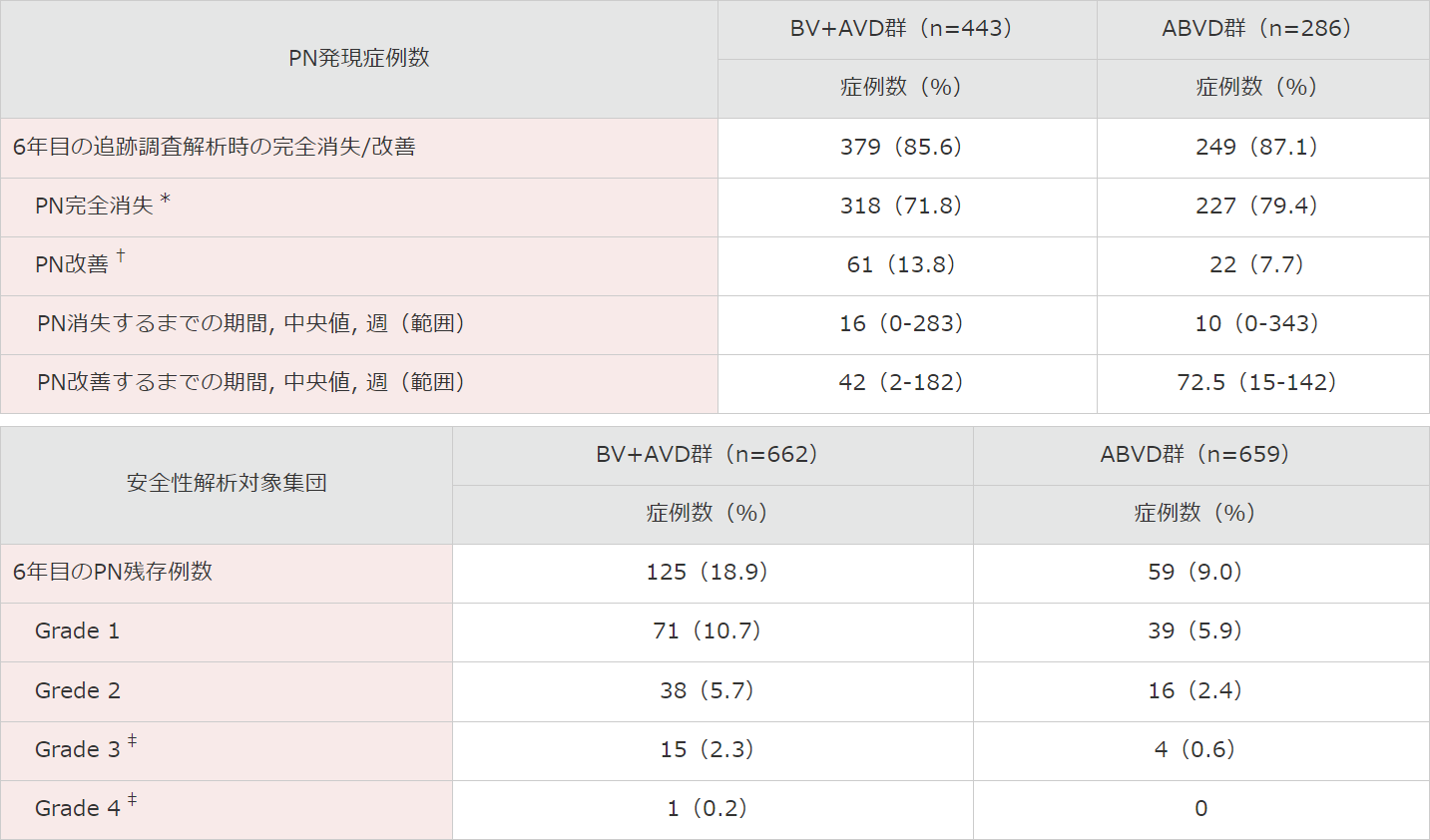
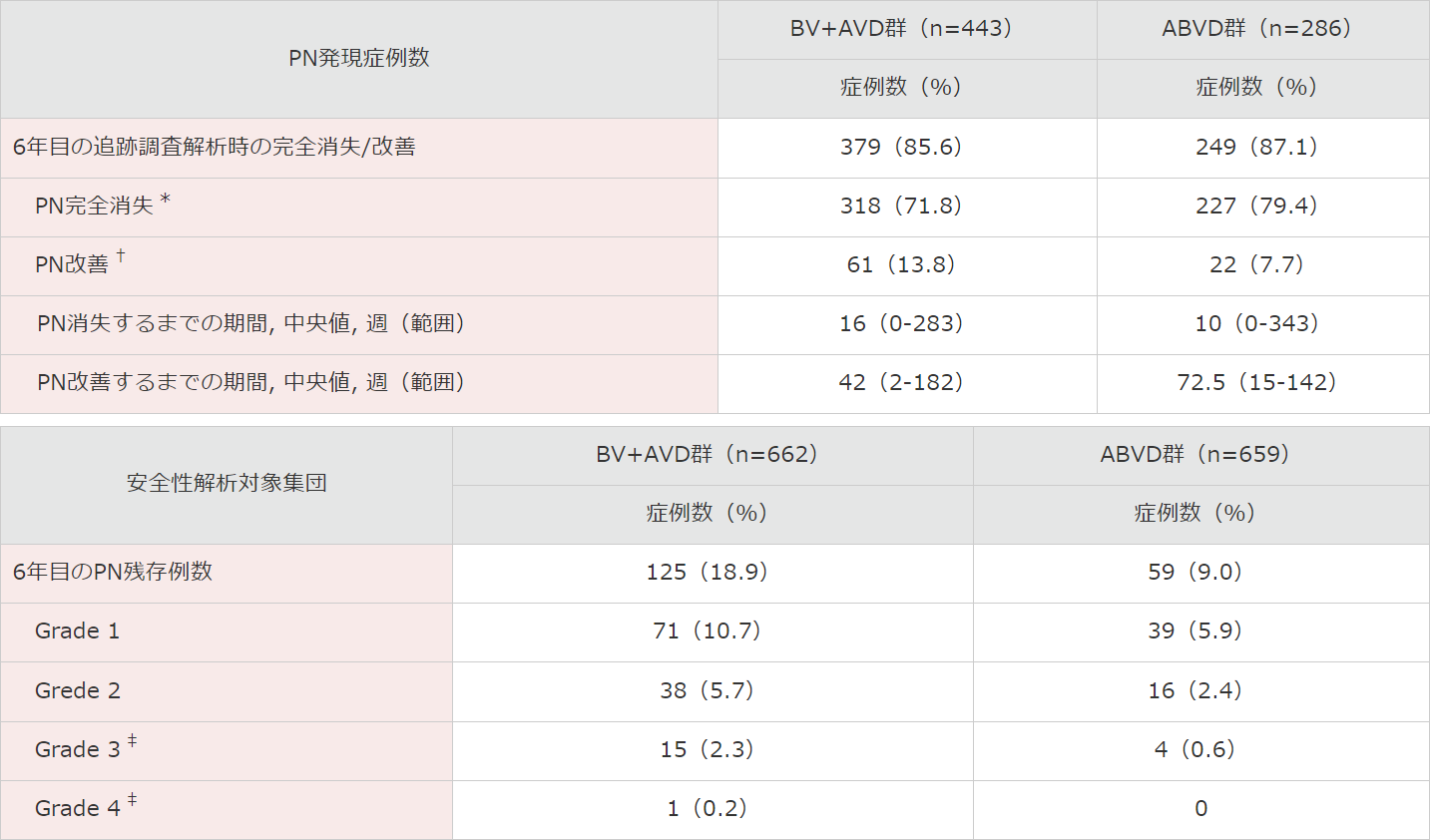
■ 末梢神経障害(PN)の発現状況注)(安全性解析対象集団)
末梢神経障害は、BV+AVD群の662例中443例(67%)及びABVD群の659例中286例(43%)に発現しました。
最終追跡調査時点で、BV+AVD群では379例(85.6%)が完全消失[318例(71.8%)]又は改善[61例(13.8%)]に至りました。ABVD群では249例(87.1%)が完全消失[227例(79.4%)]又は改善[22例(7.7%)]に至りました。
最終追跡調査時点において、BV+AVD群の662例中125例(18.9%)、及びABVD群の659例中59例(9.0%)に、PNの持続が認められました。
大部分の事象はGrade 1[BV+AVD群の71例(10.7%)、ABVD群の39例(5.9%)]又はGrade 2[それぞれ、38例(5.7%)、16例(2.4%)]でした。
持続するGrade 3又は4のPNの評価は、BV+AVD群の16例中11例(3例は死亡、4例は追跡不能、4例は症状の消失又は改善前に試験から脱落)、及びABVD群の4例(2例は死亡、2例は追跡不能)に関しては限定的でした。
2回目の中間解析
* 消失は、後遺症の有無に関わらず消失/改善した、又は既存の事象の最新の評価時点で、ベースラインかそれ以下の重篤度に戻ったものとして定義された。
† 改善は、消失又は最も重篤なGradeから1 Grade以上の重篤度の低減を認め、その後にそれより高いGradeとならないこととして定義された。
改善までの期間は、最重篤グレードの発生日から、その後にそれを上回るGradeになることなく毒性Gradeが最大Gradeを下回った最初の日までとして定義された。
‡ 消失又は改善の前に追跡不能又は死亡となった被験者は打ち切りとしなかった[BV+AVD群:11/16例(Grade 4のPNを認めた1例を含む)、ABVD群:4/4例]。
注)その他の副次評価項目である安全性に関して、ブレンツキシマブ ベドチンの医薬品リスク管理計画の【重要な特定されたリスク】の一つであるPNを取り上げました。
* 消失は、後遺症の有無に関わらず消失/改善した、又は既存の事象の最新の評価時点で、ベースラインかそれ以下の重篤度に戻ったものとして定義された。
† 改善は、消失又は最も重篤なGradeから1 Grade以上の重篤度の低減を認め、その後にそれより高いGradeとならないこととして定義された。
改善までの期間は、最重篤グレードの発生日から、その後にそれを上回るGradeになることなく毒性Gradeが最大Gradeを下回った最初の日までとして定義された。
‡ 消失又は改善の前に追跡不能又は死亡となった被験者は打ち切りとしなかった[BV+AVD群:11/16例(Grade 4のPNを認めた1例を含む)、ABVD群:4/4例]。
注)その他の副次評価項目である安全性に関して、ブレンツキシマブ ベドチンの医薬品リスク管理計画の【重要な特定されたリスク】の一つであるPNを取り上げました。